







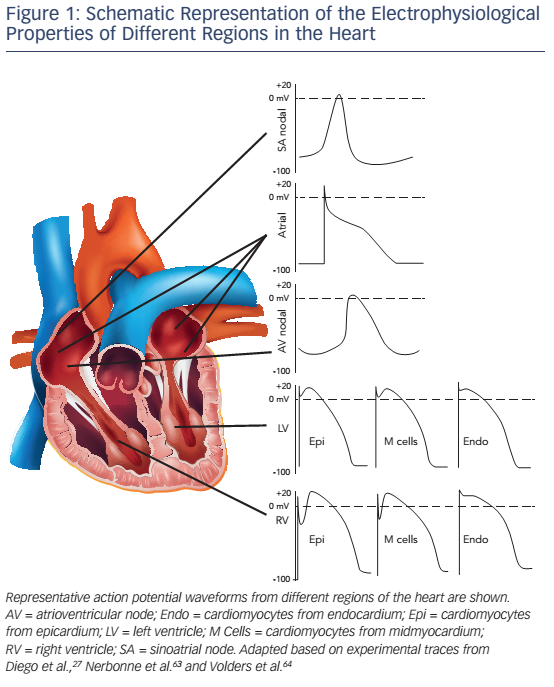
Optimal cardiac function depends on appropriate rate and force of contraction, with specific cardiac regions having developed particular beat-to-beat properties depending on their individual functions. For example, isovolumetric contraction time is shorter in the right ventricle (RV) than in the left ventricle (LV). At the cellular level, cardiac function is regulated by regional cardiomyocyte electrophysiological and Ca2+- handling properties (see Figure 1). Differences in these properties between nodal cells and working myocardium,1,2 atrial and ventricular cardiomyocytes1,3,4 and different layers of the LV wall (endo-, mid- and epicardium)5–7 have been well established. Although electrophysiological differences between left and right sides of the heart have been less extensively characterised there is evidence for clinically relevant left-toright differences in the atrium1,8–10 and the ventricle.1,5,11–14 Here, we review the known differences in LV and RV electrophysiology and Ca2+ handling at baseline and during pathophysiological conditions. Furthermore, we discuss the implications of these differences for arrhythmogenesis.
Basic Cardiac Electrophysiology and Arrhythmia Mechanisms
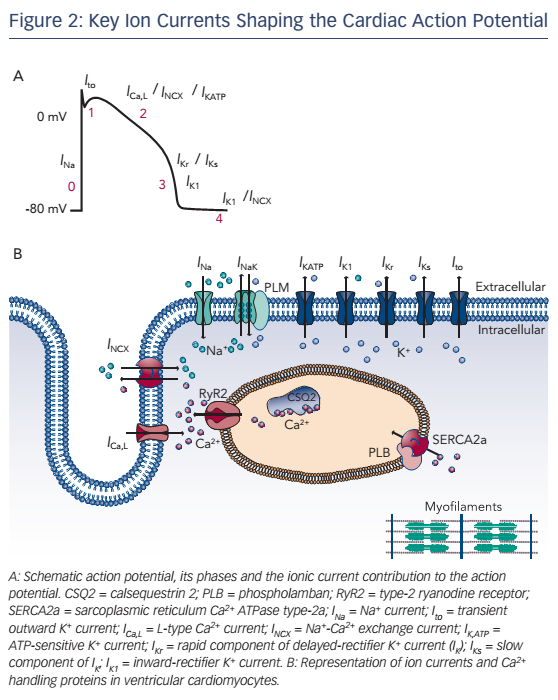
Cardiac excitation–contraction (EC) coupling is a sequence of events occurring in cardiomyocytes upon electrical activation, resulting in the generation of an action potential (AP) and subsequent cardiomyocyte contraction (see Figure 2). This sequence shows many similarities between different cell types, notably between LV and RV cardiomyocytes. In this section we briefly summarise the common features.
EC coupling involves an initial depolarisation of the membrane potential due to activation of Na+ channels and consequent opening of voltage-dependent K+ channels and L-type Ca2+ channels. The K+ channels consist of delayed-rectifier channels with distinct kinetics, underlying a transient-outward K+ current (Ito), as well as rapid and slow delayed-rectifier K+ currents (IKr and IKs, respectively). These currents play a major role in the AP repolarisation and critically determine AP duration (APD). The inward-rectifier K+ current (IK1) activates late during the AP and controls final repolarisation and resting membrane potential stability. L-type Ca2+ channels activate early during the AP and provide a depolarising current (ICa,L). Although the current subsequently declines due to voltage- and Ca2+-dependent inactivation, it supports the plateau phase of the ventricular AP (see Figure 2A). Moreover, the Ca2+ entering the cardiomyocyte through L-type Ca2+ channels plays a critical role initiating EC coupling by activating type-2 ryanodine receptor (RyR2) channels on the sarcoplasmic reticulum (SR) membrane, producing a much larger SR Ca2+ release. This process is termed Ca2+-induced Ca2+ release (CICR) and results in an increase in the cytoplasmic Ca2+ concentration sufficient to activate the contractile apparatus, initiating cardiomyocyte contraction.15 Subsequently, resequestration of Ca2+ in the SR by the SR Ca2+ ATPase type-2a (SERCA2a) and extrusion of Ca2+ to the extracellular space by the Na+-Ca2+ exchanger type-1 (NCX1) returns cytosolic Ca2+ to diastolic levels, promoting cellular relaxation. Finally, ionic homeostasis of intracellular Na+ and K+ is maintained by the Na+/K+-ATPase and the resulting current (INaK) contributes to membrane repolarisation and stability of the resting membrane potential.
Cardiac arrhythmias can arise when normal impulse generation or impulse propagation is compromised.16 Abnormal impulse formation outside of the sinoatrial node (ectopic activity) generally results from instabilities of the membrane potential during or after the AP (termed early or delayed after depolarisations [EADs/DADs]). EADs are promoted by excessive APD prolongation (e.g., due to loss of repolarising K+ currents), resulting in ICa,L reactivation and secondary depolarisations.17 DADs, on the other hand, result from spontaneous SR Ca2+-release events that activate NCX1. Since NCX1 is electrogenic (exchanging one Ca2+ for three Na+), this produces a transient inward current and depolarisation of the membrane potential.18–20
When EADs or DADs of sufficient amplitude occur synchronised between a large enough number of cells, the electrical activity can propagate through the remainder of the myocardium as ectopic (triggered) activity. Impulse propagation is mainly determined by electrical cell-to-cell coupling through gap-junction channels, presence of non-conducting tissue (non-excitable cells, fibrosis), and the local source/sink balance (e.g., depending on INa availability). Slow, heterogeneous conduction and short effective refractory periods promote reentrant activity, the predominant arrhythmia maintaining mechanism.21,22 Both ectopic activity and reentry are promoted by electrical, structural and neurohumoral ventricular remodeling, occurring in both hereditary and acquired cardiovascular diseases.
Differences Between Left Ventricle and Right Ventricle Cellular Electrophysiology at Baseline and During Pathophysiological Remodeling
Differences in Ion Channel Properties
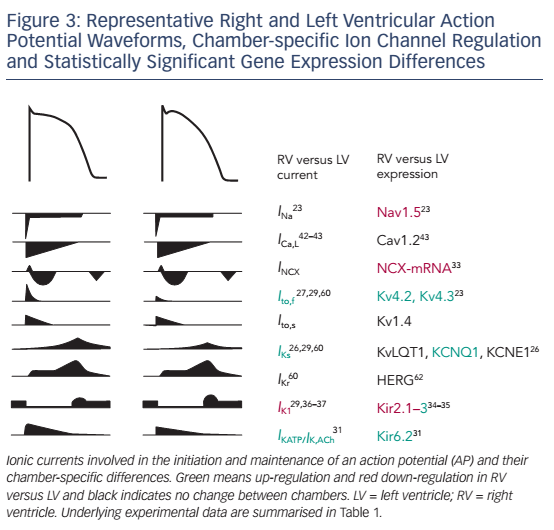
The AP is generated by specific voltage-gated ion currents so it is logical that electrophysiological differences between heart chambers result in large part from differences in ion currents (see Figure 3).1 Indeed, electrophysiological specialisation of different regions of the heart has resulted in characteristic AP patterns for each region (see Figure 1).6
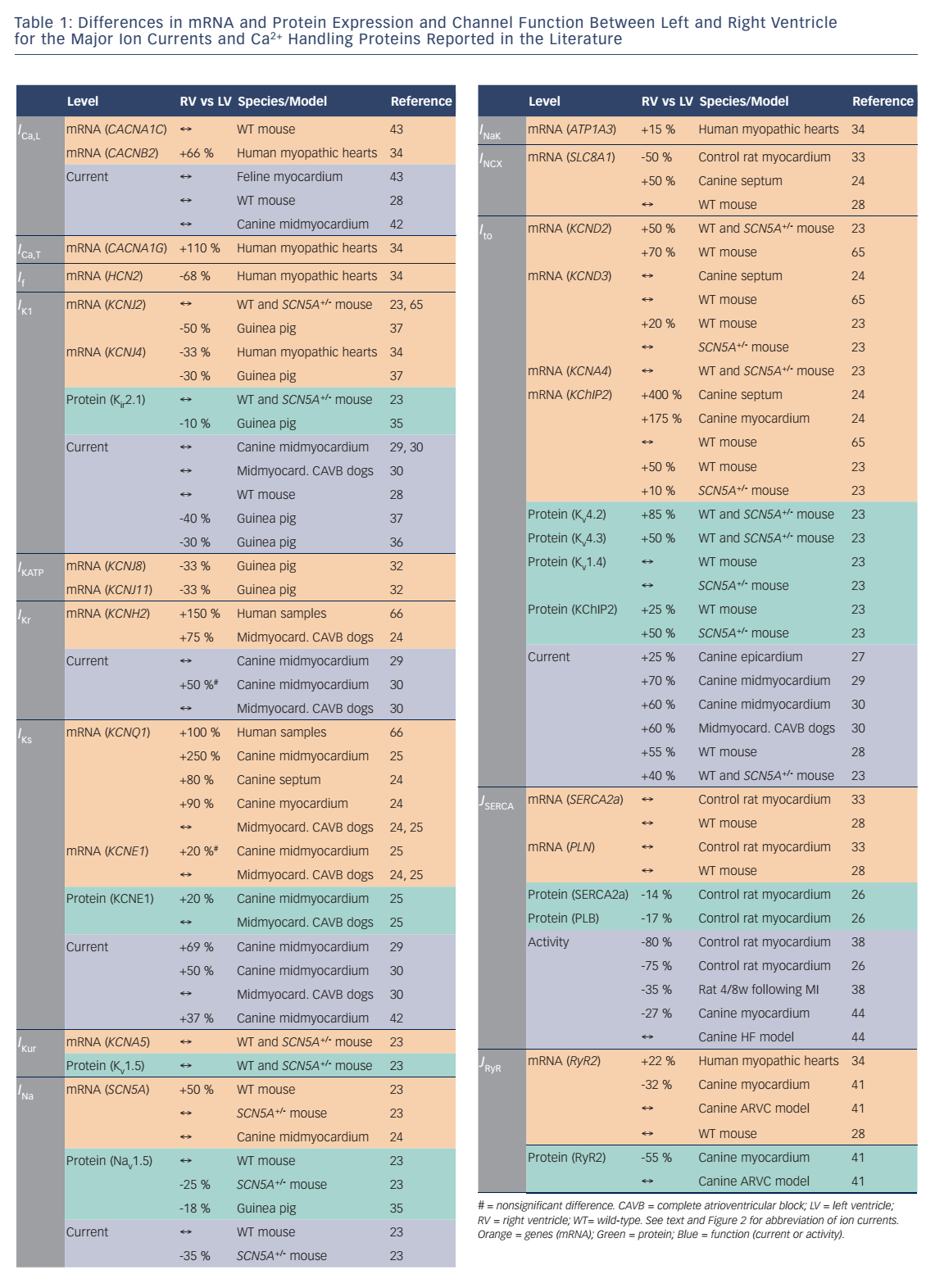
Potential ionic differences between basal LV and RV cellular electrophysiology have been identified at the mRNA, protein and functional levels (see Table 1). In most species and experimental models, the RV myocardium shows a relative overexpression of Kv4.2, Kv4.3 and KChIP2,23,24 molecular components of Ito, as well as greater KCNQ1 expression,25,26 part of the IKs macromolecular complex. In agreement, a number of studies observed larger IKs and Ito in RV compared with LV.27–30 In addition, some studies have observed changes in the gene expression of Kir6.1/Kir6.2, underlying the ATP-sensitive K+ current (IKATP),31,32NCX133 and K ir2.1/Kir2.3, molecular components of IK1.34,35 Consistent with these molecular data, IK1 density is larger in LV myocytes from guinea pigs, contributing to the stabilisation of the high-frequency rotors in LV.36,37 However, other studies in different animal models did not find a significant difference between LV and RV IK1.28,29 Finally, some studies have suggested that INa might be smaller in RV than LV.23,35
At the cellular level, APs showed deeper notches, shorter APDs at 50 % and 95 % of repolarisation and less APD prolongation on slowing of the pacing rate in RV than LV,27,24,29 consistent with the larger Ito and IKs. Similarly, duration of monophasic APs in vivo was shorter in RV than in LV.25 Resting membrane potential and AP upstroke velocity did not differ between LV and RV in these studies.27,29
Although these results clearly suggest different electrophysiological phenotypes of the RV and LV, there is significant disagreement between the different species and experimental settings, as well as between expression data and functional studies. Ito is a notable exception being consistently larger in RV than LV (see Table 1). Furthermore, the role of individual electrophysiological differences in chamber-specific proarrhythmia is largely unknown. Similarly, only a limited number of studies have investigated whether chamber-specific electrical and structural remodeling processes regulate these differences between both ventricles. Volders et al.30 reported an RV-specific downregulation of IKr and a disappearance of the LV/RV differences in IKs in a dog model with chronic complete atrioventricular block. These findings were confirmed at the transcriptional level by downregulation of KCNH2 and KCNQ1 expression (underlying IKr and IKs, respectively) in subsequent studies.24,25 Reduction of repolarisation reserve due to K+-channel downregulation is linked to an increased risk of ventricular arrhythmias and sudden cardiac death in this experimental model. Differences in Ito between LV and RV, on the other hand, remained intact in this model, highlighting the complexity of chamber-selective and channel-specific remodeling.
Differences in Ca2+ Handling and Contractility
Interventricular differences in Ca2+ handling and contractility have been predominantly investigated in rodents (see Table 1). No intrinsic RV/LV differences were found in gene expression of SERCA2a, its inhibitory regulator phospholamban (PLB), RyR2, NCX1 or the pore-forming α subunit of the L-type Ca2+ channel.28 Similarly, SR Ca2+ uptake was not different between both ventricles. Nonetheless, systolic [Ca2+] and cell shortening were larger in LV than RV. AP clamp experiments showed that the observed interventricular differences in Ca2+ handling were due to differences in AP morphology, with shorter APD in the RV compared with the LV, affecting ICa,L-mediated Ca2+ influx.28 SERCA2a and PLB mRNA levels were also similar in both ventricles in rats,33 whereas protein expression of both was lower in RV.26 In accordance, SR Ca2+ sequestration was slower in RV compared with LV in normal rat myocardium,38,26 and Ca2+-transient decay was slower in RV.26 There were no interventricular differences in diastolic or systolic [Ca2+] but cell shortening was smaller in rat RV cardiomyocytes. Furthermore, both ventricles showed opposite changes in SR Ca2+ sequestration upon induced myocardial infarction. While in the LV Ca2+ uptake decreased, it increased in RV, affecting the rate of relaxation and contraction. This suggests that failure of the LV promotes differential RV remodeling and potentially proarrhythmic chamber dyssynchrony.38
There are important differences in electrophysiology and Ca2+ handling between rodents and larger mammals (including humans). Rodentsrely heavily on SR Ca2+ cycling, with >90 % of the total Ca2+ flux during a single beat resulting from SR Ca2+ release and subsequent SR Ca2+ reuptake. By contrast, in larger mammals there is a much larger role for Ca2+ entry via ICa,L and NCX1-mediated Ca2+ extrusion, which account for ~30 % of the total Ca2+ flux.39,40 Thus, extrapolation of the data on LV/RV differences in Ca2+ handling from rodents to humans is difficult.
There are few data available about chamber-specific Ca2+-handling properties in large mammals. RyR2 mRNA and protein expression were lower in RV compared with LV in myocardium of control dogs.41 By contrast, RyR2 gene expression was larger in RV in ventricular samples from cardiomyopathy patients.34 At the functional level, no differences in basal Ca2+-transient amplitude or sarcomere shortening could be detected between RV and LV in canine cardiomyocytes.42 Cardiomyocyte shortening and relaxation rate in RV and LV were also similar in cats.43 Interestingly, interventricular differences in RyR2 expression were eliminated, and total RyR2 expression decreased in dogs with arrhythmogenic right ventricular cardiomyopathy.41 Similarly, Gupta et al.44 found reduced SERCA2a activity and protein levels in LV, but not RV, in dogs with chronic heart failure, eliminating interventricular differences. These data suggest that interventricular differences in Ca2+ handling are species dependent and can be further regulated by chamber-specific disease-related remodeling.
Interventricular Differences in the Regulation of Cardiomyocyte Electrophysiology and Ca2+ Handling
Numerous studies have highlighted the importance of posttranslational regulation of ion channels and Ca2+-handling proteins to control cardiac electrophysiology and contractility in response to various neurohumoral conditions.15,45–47 Activation of β-adrenoceptors with isoprenaline similarly regulates ICa,L and IKs in canine LV and RV cardiomyocytes, whereas it increased sarcomere shortening 10-fold versus 25-fold and Ca2+-transient amplitude two-fold versus threefold in LV versus RV cardiomyocytes, respectively, highlighting clear interventricular differences in the regulation of cardiomyocyte Ca2+ handling.42 These differences were found to be due to a selective isoprenaline-induced increase in cytoplasmic cAMP in RV, resulting from distinct rates of cAMP degradation by type-3 and type-4 phosphodiesterases.42 By contrast, Ca2+/calmodulin-dependent kinase type-II (CaMKII)-dependent phosphorylation of RyR2, SERCA2a and PLB following application of exogeneous calmodulin/Ca2+ was reduced in RV versus LV myocardium of rats,26 thus suggesting potential interventricular differences in CaMKII signaling.
The RV and LV also showed opposite inotropic responses to α1-adrenergic stimulation,48 which was at least in part due to heterogeneous effects on LV/RV intracellular Ca2+ handling.49 Finally, β2-adrenoceptors were found highly upregulated in LV, but not RV, in rats with chronic mild stress.50 Thus, although relatively little is known about interventricular differences in ion channel regulation, presently available data suggest a complex system with chamber-specific remodeling of pre-existing interventricular differences in regulatory signaling pathways, which act upon differences in basal LV versus RV electrophysiology and Ca2+ handling.
Mechanisms Underlying Left Ventricle versus Right Ventricle Differences
The electrophysiological differences between the LV and RV can at least partially be attributed to the distinct embryological origin of the LV, arising from the first heart field, and the RV, arising from the second heart field.5,51 Furthermore, within the RV there are embryological differences between the RV free wall and the outflow tract, with the latter forming at a later stage during development.52 Each developmental origin is associated with expression of different transcription factors.5 For example, Hand1 is predominantly found in the first heart field, and Hand2 in the second heart field. Similarly Tbx2 is specifically found in the outflow tract of the embryonic heart.52 Although the exact factors regulating mRNA expression of each ion channel remain largely unknown, the distinct expression profiles of ion channels and Ca2+ handling proteins in the LV and RV (see Table 1) strongly suggest a role for chamber-specific transcriptional regulation. Quantitative differences between mRNA, protein and current levels in LV versus RV suggest other potential forms of regulation, which may include transcriptional regulation of regulatory subunits or other components of the macromolecular ion-channel complex; microRNAdependent regulation of protein levels; differences in trafficking, membrane insertion or degradation; distinct subcellular localisation or post-translational modification.53,1,45,54
Clinical implications
Due to its unique geometry and cell biology the RV behaves differently from the LV in a variety of pathophysiological conditions and deterioration of right ventricular function strongly predicts clinical outcomes in a variety of circumstances.13,55 In addition to these structural aspects, Brugada syndrome (BrS) provides an example of the relevance of interventricular electrophysiological differences for arrhythmogenesis. BrS is characterised by right-precordial ST-segment elevation on the body-surface electrocardiogram (ECG) and is associated with an increased risk for sudden cardiac death due to malignant ventricular tachyarrhythmias.56,57 It was traditionally considered a congenital channelopathy in the absence of overt structural heart disease, linked predominantly to loss-of-function mutations in the SCN5A gene (locus 3p21) encoding the pore-forming α subunit of the Na+ channel. However, recent work has demonstrated the greater complexity of the disease, with at least 18 other genes as well as acquired functional and structural abnormalities also implicated. 58,57
Two arrhythmogenic mechanisms have generally been proposed for BrS.57,59 In the repolarisation disorder hypothesis, the loss of INa in combination with a large Ito in the RV epicardium, particularly near the RV outflow tract, results in a local loss of AP spike-and-dome morphology and pronounced regional APD shortening, producing ST-segment elevation in the right-precordial leads. The resulting repolarisation gradient could predispose to ventricular arrhythmogenesis via phase-2 reentry. 60 The depolarisation hypothesis, on the other hand, is based on delayed activation of the RV outflow tract, resulting in large potential gradients that produce the ST-segment elevation.
Recent work in post-mortem hearts with familial BrS indeed found evidence for increased local levels of fibrosis and reduced levels of gap-junction proteins (notably connexin-43) in the RV outflow tract,61 supporting a role for region-specific structural abnormalities and conduction disturbances in BrS. A mouse model with heterozygous knock-out of SCN5A has also suggested that the RV might be particularly sensitive to loss of functional Na+ channels, with a larger reduction in INa in RV compared with LV.23 Similarly, Veeraraghavan and Poelzing35 showed that heterogeneity in Nav1.5 expression in guinea pig may become a significant determinant of conduction heterogeneities under conditions where INa is functionally reduced. However, this study also highlights that conduction heterogeneities can be further modulated by interventricular differences in other ion channels, including IK1 .35 Indeed, recent non-invasive eletrocardiographic imaging of BrS patients revealed both slow, discontinuous conduction and steep repolarisation gradients in the RV outflow tract, suggesting interactions between both mechanisms.14 Thus, regardless of the exact mechanism (depolarisation versus repolarisation), RV-specific electrophysiological and structural properties play a critical role in the phenotypic presentation of BrS patients.
Besides BrS, interventricular electrophysiological differences may play a role in ventricular arrhythmogenesis in a variety of conditions. In general, steep repolarisation gradients have been considered proarrhythmic, and interventricular differences in ion-channel expression, regulation or disease-related remodeling may contribute to such gradients.5 For example, interventricular differences in IKATP could be an important determinant of LV/RV APD gradients during global ischaemia,32 and heterogeneous ventricular chamber responses to hypokalaemia and IK1 blockade contributed to bifurcated T-wave patterns in guinea pig.62 Similarly, differential downregulation of RV and LV delayed rectifier K+ currents could contribute to repolarisation abnormalities and arrhythmogenesis in patients with cardiac hypertrophy or failure.30
Conclusion
Chamber-specific heterogeneity in cardiac electrophysiology is a physiological phenomenon, which contributes to fine-tuning of cardiac function. During the last two decades some studies have started to identify differences in ion channel expression and function between RV and LV. However, only limited information is available about the distinct remodeling of each ventricle and the subsequent impact on cardiac arrhythmogenesis. This holds particularly true for post-translational modifications affecting channel function and cardiomyocyte Ca2+ handling. Further extensive work, ideally in human samples or large animal models, is needed to define the precise role of interventricular electrophysiological differences in ventricular remodeling, cardiac dysfunction and arrhythmogenesis.