









Long QT Syndrome
Long QT syndrome (LQTS) is a potentially severe arrhythmogenic disorder, affecting more than one in 2,000 people worldwide.1 It is characterised by a marked prolongation of the QT interval on the electrocardiogram and major cardiac events, such as syncope, cardiac arrest or sudden death, especially under conditions of physical or emotional stress.2,3 The current diagnostic criteria and options for effective management have been recently reviewed.4 As the first manifestation of the disease is frequently a lethal arrhythmic event, early diagnosis and therapy are extremely important and recent advances in genetics offer new opportunities.3 Besides its central role for the discovery of the molecular basis of LQTS and for the identification of the first gene-specific therapy, genetics became pivotal for the identification of the disease-causing genes and mutations in routine diagnostic practice and for gene-specific management.3–5 However, the low and variable penetrance exhibited by LQTS and the many confounding factors represent challenges to diagnosis and obstacles for the implementation of successful proactive treatments.6 In this context, multiple groups including ours have started to investigate and identify factors – either genetic, pharmacological or environmental – able to modify the clinical course of the disease by acting as modifiers of gene/protein function, expression or regulation.7 People with identical pathogenic mutations, even members of the same family, may display different degrees of clinical severity, symptoms or even be silent carriers of the mutation, which complicates the possibility of having unambiguous predictions of disease phenotypes simply based on the presence of disease-causing mutations in LQTS genes or when someone has a family history of the disease.7–12
Diagnosis of LQTS may also be hindered by the presence of uncharacterised rare variants; given that the pace by which modern genetic tools can extract huge amounts of information is unprecedented, the field is progressively accumulating a growing number of rare genetic variants associated with LQTS subpopulations; these, initially defined variants of unknown/uncertain significance (VUS), are variants short-listed in the genetic analysis but for which evidence of being benign or pathogenic does not allow to draw univocal conclusions.13,14
The complexity behind some of the puzzling LQTS clinical manifestations has still to be fully unveiled and this leaves many patients, who might need clinical attention during the course of their lives, undiagnosed and unprotected. Therapies should be personalised for these subjects, but current tools and knowledge do not allow sufficient predictivity or mutation-specific remedies. To study such a high complexity, the need for valid and reliable experimental models intensifies; traditional models as heterologous systems are incapable of completely recapitulating genetic and phenotypic features, while animal models, particularly in rodents, do have electrophysiological characteristics that are often species-specific and very different from humans.15 This hampers the possibility of promptly translating results obtained in vitro directly to the clinics, with significant delays occurring between the discovery of promising treatments and their clinical application. For all these reasons, new experimental models, more predictive of human physiology and less limited by costs and ethics, are required and cardiomyocytes (CMs) derived from induced pluripotent stem cells (iPSC-CMs) have emerged in the past few years as a leading platform for these studies.
Almost 10 years after the first LQTS model with human iPSC-CMs (hiPSC-CMs) was published, we will briefly discuss the state of the art, some of the obstacles that need to be solved and the fascinating transition of hiPSC-CMs as precision medicine tools that are used to preliminarily investigate experimental therapies and guide pharmacological treatments.16 We will focus on the main forms of LQTS.
Human-induced Pluripotent Stem Cells
In 2006, Shinya Yamanaka and Kazutoshi Takahashi discovered that somatic cells could be reprogrammed to a pluripotent state by transfecting four factors – OCT4, SOX2, KLF4, cMYC – subsequently called Yamanaka factors.17,18 These reprogrammed cells, called iPSCs, can be derived from virtually anyone and represent an unprecedented source of human-based material that can be differentiated into multiple cell types through tailored differentiation protocols, including cardiomyogenic differentiation.19 Originally, iPSCs were derived from skin fibroblasts, which requires a biopsy, often difficult to propose for young children or to vulnerable subjects often already daunted by life-threatening diseases. Thus, new methods have been developed to obtain somatic cells from more accessible and less invasive tissues samples such as blood or urine.20,21
In addition to the obvious electrophysiological and molecular advantages of being of human origin, these cells share genotypes with their donors; this feature makes iPSCs candidate experimental models to study diseases of genetic origin, with LQTS and cardiac channelopathies being among the first to be reproduced with this technology. In 2010, the pioneering work of Alessandra Moretti et al., demonstrated that hiPSC-CMs could become powerful tools to study the functional effects of ion channel mutations identified in LQTS patients.16 However, as the molecular and functional characteristics of hiPSC-CMs are still far from those of adult CMs, multiple protocols were invented to promote their maturation.22 These strategies include the use of prolonged cultures, chemically-defined media, the generation of tridimensional structures tailored for electrical stimulation protocols or co-cultures of CMs with other relevant cell types.23–31 Overall, these experimental models are progressively improving and the techniques to study the phenotype of hiPSC-CMs have advanced in parallel.
Methods for the study of Long QT Syndrome with Human-induced Pluripotent Stem Cells
The in vitro investigation of LQTS has the primary endpoint to ascertain whether and how the CMs’ electrophysiology has been modified as a consequence of LQTS-causing gene mutations. The patch clamp technique is the reference platform of choice to precisely quantify the phenotype of hiPSC-CMs. Although limited by its low throughput and disruptiveness, patch clamp allows quantitative recordings of action potentials (AP) and ion currents; furthermore, the combination of real-time computational ion current simulations with in vitro experiments has merged in the dynamic clamp technique.32 Although not particularly new at the time hiPSC-CMs were first derived, it was embraced to compensate the intrinsically reduced inward rectifier potassium current (IK1) magnitude, typical of isolated hiPSC-CMs,33–35 with an artificial IK1 injected in real-time. This technique transforms the action potential (AP) of hiPSC-CMs into one with more mature features as greater upstroke velocity and the presence of the AP notch, allowing voltage-gated ion channels to properly work as they were in more physiological conditions; this proved particularly relevant to capture the electrophysiological alterations caused by LQTS pathogenic mutations.33,34
However, since the heart is a functional syncytium, measurements of the downstream effects of LQTS mutations have greater translational power when performed on multicellular networks. At sufficient density, hiPSC-CMs do form functional beating mono- or multilayered sheets, which can be scrutinised with the multielectrode array (MEA) technology. Originally developed to study neuronal circuits, MEAs have gained interest in recent years with the development of stem cell-based cardiac disease models.36 Furthermore, the MEA technology has also been defined as one of the platforms of choice in the context of the US Food and Drug Administration’s Comprehensive in vitro Proarrhythmia Assay (CiPA), a task group which aims to identify the most suitable platform and protocols for drug screening and safety pharmacology.37
Other technologies with low invasiveness have been implemented as chemical or genetically-encoded voltage or calcium indicators, optogenetic actuators or imaging algorithms to investigate specific parameters of the diseased phenotype as a contraction and they all became widely used to obtain functional information with medium-high throughput.38–44
Long QT Syndrome Subtypes Studied with Human-induced Pluripotent Stem Cell Cardiomyocytes
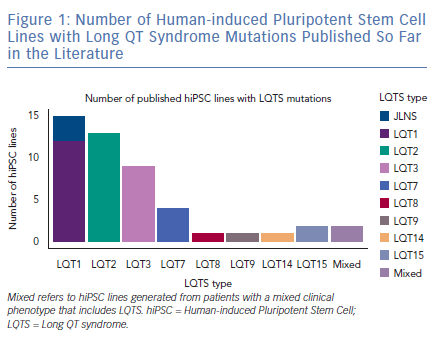
As expected by the high prevalence in patients, the first three forms of LQTS are also those primarily modelled in vitro.1 However, other LQTS types have been identified and successfully reproduced with hiPSC-CMs (Figure 1); we will focus on these three main types and on the extremely severe forms related to mutations on the genes encoding for calmodulin (CaM). Common in vitro phenotypes across all LQTS subtypes include prolonged AP durations (APDs), measured with patch clamp or voltage-sensitive indicators, prolonged field potential (FP) durations (FPD) measured with MEAs or prolonged Ca2+ and contraction transients (Figure 2). Some of the cell lines also experienced arrhythmogenic events as delayed after depolarisations (DADs) and early after depolarisations (EADs) in baseline conditions or after a pharmacological challenge.36
Long QT Syndrome Type 1 and Jervell and Lange-Nielsen Syndrome
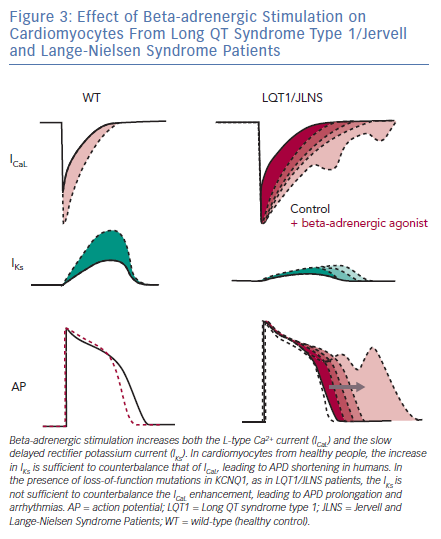
LQTS type 1 (LQT1) is the most prevalent form of congenital LQTS, accounting for approximately 40–55% of all cases and its heterozygous form, called Romano-Ward syndrome, was the first LQTS type to be recapitulated with hiPSC-CMs.45–48 The severe homozygous form, when associated with deafness, is defined as Jervell and Lange-Nielsen syndrome (JLNS).49,50 People with LQT1 and JLNS are affected by mutations in KCNQ1 that induce loss of functions in Kv7.1, an important potassium channel responsible for the slow delayed rectifier potassium current (IKs). Although IKs is not straightforward to record in hiPSC-CMs due to their limited cell capacitance, which reduces the total current density, and to a late IKs onset during CM development,51 partially mitigated by certain maturation strategies,25 in vitro models of LQT1/JLNS were able to recapitulate the pathological phenotype and exhibited marked reductions in IKs magnitude when compared to hiPSC-CMs from healthy donors or to isogenic controls52–54; detailed investigation of IKs with patch clamp also revealed the possibility for hiPSC-CMs to perfectly discriminate between homozygosity and heterozygosity.51–55 The similarities between hiPSC-CMs and clinical disease features extend also to triggers of arrhythmias; as previously described in humans, an increased activation of the sympathetic cascade is a clear arrhythmogenic trigger for LQT1/JLNS.56 When this is reproduced in vitro by stimulating hiPSC-CMs with beta-adrenergic agonists, their effect on L-type Ca2+ current outweighs the repolarisation capacity of the limited IKs magnitude available in LQT1/JLNS cells, leading to APD prolongation, abnormal Ca2+ transients and arrhythmogenic events (Figure 3).
Long QT Syndrome Type 2
LQTS type 2 (LQT2) is the second most common form of LQTS, accounting for 30–45% of cases.45 It is characterised by mutations in the KCNH2 gene, which encodes for the human ether a-go-go-related gene (hERG, Kv11.1), a potassium channel responsible for the rapid delayed rectifier potassium current (IKr), pivotal for the cardiac repolarisation phase.46 LQT2 mutations may induce loss of function in IKr through four main mechanisms.57 The in vitro phenotype in hiPSC-CMs is generally clear, with marked reductions in IKr coupled to consistent APD prolongation.58–68 Clinical LQT2 triggers of arrhythmias include sudden auditory stimuli or loud noises and while these cannot be properly mimicked in vitro, alternatives as a stimulation with positive inotropes, alone or in combination with IKs blockers, are a widely used strategy to exacerbate arrhythmias in these cells.56,69
Since the majority of LQT2 mutations affect IKr through defective protein trafficking with hERG channels incapable of maturing and reaching the CMs’ membrane, researchers have adopted several strategies to restore proper hERG expression or function.70 Some rescued the LQT2 phenotype with compounds able to increase the conductance of wild type hERGs, other with proteasome inhibitors59,61,67 or, more recently, through a drug repurposing strategy which promoted the maturation of mutant hERGs in vitro.58,62 This drug repurposing strategy was very rapidly translated to the two LQT2 patients whose iPSC-CMs were previously tested in vitro and proved that, besides the expected discrepancies between the clinical and in vitro conditions, the data obtained in patient-specific iPSC-CMs do offer a certain degree of predictivity in the clinical situation.71

Long QT Syndrome Type 3
LQTS type 3 (LQT3) is the third most common form of LQTS, accounting for 5–13% of all congenital LQTS cases.45,46 It is the consequence of gain-of-function mutations in the SCN5A gene, leading to an increase in the cardiac sodium current (INa); different biophysical changes induced by LQT3 mutations may increase either the peak INa or also affect its late component(INa,L), the latter involved in electrophysiological abnormalities in multiple tissues, such as pancreatic beta-cells, cardiomyocytes or neurons.72,73 Despite INa overall current magnitude is lower in hiPSC-CMs compared with adult CMs and published hiPSC-CM models of LQT3 did correctly recapitulate increases in INa and other features of LQTS as EADs or prolonged calcium transients; APD was prolonged in the majority of models even though some authors described larger than ideal inter-line and inter-cell phenotypic variability.66,74–77 Furthermore, hiPSC-CMs with LQT3 mutations reproduced the expected effect of pharmacological therapies based on sodium channel blockers, which were able to rescue the prolonged APD and FPD and to suppress arrhythmogenic events in a dose-dependent way.74,76,78 Of note, some authors were also able to model through hiPSC-CMs some cardiac disorders with overlapping phenotypes of LQT3 and Brugada syndrome.75
Long QT Syndrome Type 14 and 15
LQTS types 14 (LQT14), 15 (LQT15), and the upcoming LQTS type 16 (LQT16), which is not yet officially approved, are characterised by mutations in one of the three genes (CALM1–3) encoding for CaM.79 Discovered by our group as associated with severe forms of LQTS and later investigated with hiPSC-CMs, these mutations impair the activity of CaM by either reducing its Ca2+-binding affinity or by altering its capacity to interact with downstream targets.33,80–82 Since CaM is a ubiquitous protein, present in CMs in limited pools, the consequences are dramatic, with very young people experiencing severe arrhythmias since birth and for whom current therapies are insufficient or inappropriate.83–85 These severe phenotypic characteristics have been perfectly captured by the hiPSC technology, with hiPSC-CMs from patients with CALM mutations exhibiting pathologically slowed ICaL Ca2+-dependent inactivation and massively prolonged APDs. A phenotypic rescue has been attempted in hiPSC-CMs through novel pharmacological or biotechnological strategies, with promising results that have already obtained preliminary translation to patients.33,81,82,86
Latest Challenges
The aforementioned disease models represent a good approximation of the clinical condition and have pushed scientists to explore potential applications for hiPSC-CMs. A few of the most recent challenges of the cardiac hiPSC field will be discussed below.
Data Variability and Gene Editing
The correlation between in vitro and in vivo data is still either uncharted territory or has resulted in contradictory findings in the literature. Although evidence has demonstrated that a fair correlation between clinical and in vitro recordings exists (i.e. LQTS cell lines generally do have prolonged APD/FPD in vitro), we still do not know precisely its extent and whether and how different reprogramming or maturation strategies could modify the magnitude and the time course of the in vitro equivalents of clinically relevant parameters as the QT interval or the RR interval. In the years since the application of hiPSC technology to disease modelling, it has become clear that cell lines from different donors have innate characteristics that are independent of the specific pathogenic mutation, thus they are likely to be associated with donor-specific characteristics.87,88
Electrophysiological properties as APD, which length should directly correlate with the presence of LQTS mutations, seem cell-line specific or differentiation-method specific rather than disease-specific and the same applies to FPD-RR relationships measured with MEAs; this can be attributed to potential confounding factors as manual pipetting, cell passage number and donor’s gender or ethnicity, suggesting the need of great care when data have to be interpreted on a translational perspective.62,88,89 In this vein, one of the most important additions in the LQTS field has been the concept of isogenic lines61; gene editing techniques, either based on zinc finger nucleases (ZFN), transcription activator-like effector nuclease (TALEN) or clustered regularly Interspaced short palindromic repeats (CRISPR)/Cas9.90–93 This technology can introduce pathogenic mutations, homozygous or heterozygous, into hiPSC lines from healthy donors, or correct them without altering the cell’s genetic background. This has proved to be important for introducing more robust standards to evaluate the functional consequences of point mutations and for precision medicine approaches.12,55,61,62,82,94,95
Precision Medicine
Genetics, hiPSCs and cellular electrophysiology are progressively providing better chances of tailoring therapies to the specific mutation of the patient, and it is clear that the partnership between clinical cardiologists and basic scientists can be very fruitful96. One of the biggest advantages of hiPSC-CMs is that they share their genetic background with patients. This link can be harnessed to correlate clinically relevant information as genetic data to in vitro physiological and pharmacological responses. Patients with similar characteristics can be clustered according to their phenotype or genetic data to obtain homogenous cohorts of people who share similar characteristics. HiPSC-CMs are generated from these subjects and subsequently screened with molecular and functional assays that are able to generate data of translational relevance. Gene editing tools are used here to validate the results in the presence of identical genetic backgrounds.95 This strategy may have important consequences for risk stratification and therapeutic responses, as patients with certain VUSs would ideally be given different drug dosages based on the preliminary results of the in vitro screenings in hiPSC-CMs.
In the context of precision medicine, the identification of novel clinical applications of drugs that have already received approval from regulatory agencies (drug repurposing) has increased. When combined with hiPSCs, it allows marketed drugs that are used to treat other disorders to be tested in vitro with a remarkable throughput on differentiated cells of human origin. In this view, our group has identified that lumacaftor (LUM), a compound currently prescribed for the treatment of specific mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, associated with defects trafficking in CFTR channels, could also rescue the disease phenotype in hiPSC-CMs derived from a specific class of people with LQT2 affected by hERG trafficking problems.58 Since LUM is already marketed for the treatment of cystic fibrosis in combination with ivacaftor and its safety profile is known, we were sufficiently confident to start a clinical trial.71 The mechanisms by which LUM may rescue hERG trafficking defects are still unknown and these results will require extensive confirmation from clinical data and from other hiPSC lines from more LQT2 donors to verify whether these effects could be cell-line- or mutation-specific or whether their mechanism could be exploited for broader applications.
Conclusion
hiPSC technology has brought significant contributions to the way LQTS and cardiac arrhythmias are modelled at preclinical level97; however, more efforts are required to offer an exhaustive comprehension of the translational relevance of hiPSC findings. In our view, shared and open protocols, tighter standards from researchers, journals, funding agencies as well as the implementation of large publicly available informatic tools and datasets are the sole way to revolutionise how the iPSC technology interacts with the clinics and shift hiPSC-CMs from useful in vitro preclinical tools to something able to produce relevant information for prompt clinical decision-making. It is also clear that genetics, combined with the most recent advances in hiPSC-CMs technology, bioinformatics and artificial intelligence, will unveil future possibilities and targets for the diagnosis, risk stratification and treatment of LQTS.
Clinical Perspective
- LQTS is a potentially lethal arrhythmogenic disorder that affects more than 1 in every 2,000 children. Our knowledge of genetics in LQTS has progressively increased over the years, and we reached a mismatch between the amount of genetic information available and the functional data that allow us to interpret this genetic information.
- The emerging role of variants of unknown/uncertain significance (VUS) require in vitro experimental models capable of effectively integrating the genetic component and to extract predictive information relevant for clinical use.
- Human induced pluripotent stem cell-derived cardiomyocytes from LQTS patients, as they share the patients’ genotype, have proven to be a useful platform for in vitro investigation of the effect of pathogenic mutations of LQTS and VUS on the cardiac phenotype, and may represent the optimal tool, in combination with gene editing strategies, for large-scale investigation of the functional consequences of genetic variants.
- Large precision medicine approaches could combine the benefits of large-scale genetic studies with reliable functional readouts operating at medium to high throughput.
- Some of the current limitations of the hiPSC technology can be addressed only with combined and bidirectional efforts between clinicians and scientists.