









Since its initial description by Jervell and Lange-Nielsen in 1957,1 congenital long QT syndrome (LQTS) has been the most investigated cardiac ion channelopathy. A prolonged QT interval on the surface ECG is a surrogate measure of prolonged ventricular action potential duration (APD).
Congenital as well as acquired alterations in certain cardiac ion channels can affect their currents in such a way as to increase the APD and hence the QT interval. The inhomogeneous lengthening of the APD across the ventricular wall results in dispersion of APD, i.e. dispersion of repolarisation (DR). This, together with the tendency of prolonged APD to be associated with oscillations at the plateau level, termed early afterdepolarisations (EADs), provides the substrate of ventricular tachyarrhythmia (VT) associated with LQTS, usually referred to as torsade de pointes (TdP) VT.2
Acquired LQTS is by far, more prevalent than congenital LQTS. The vast majority of acquired LQTS is the result of the adverse effect of drugs3 and/or electrolyte abnormalities,4 which, in the majority of cases, interact with the human ether-à-go-go-related gene (hERG) encoding the pore-forming subunits (Kv11.1) of the rapidly activating delayed rectifier current, IKr. However, recent reports suggest that some drugs can also increase the late sodium current, which may contribute to their proarrhythmic effect.5
Acquired Long QT Syndrome
ECG Characteristics of Torsade de Pointes
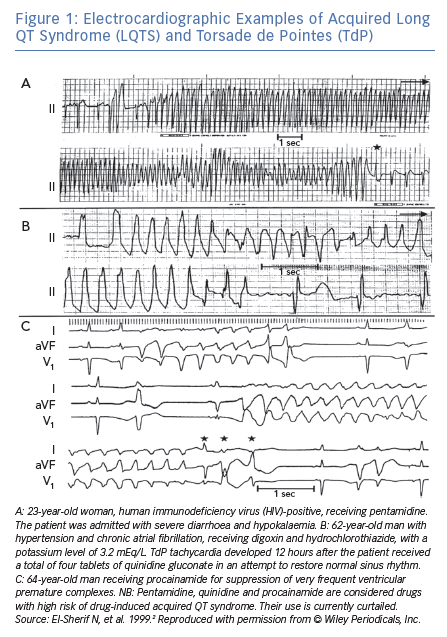
In an analysis of 150 different episodes of sustained VT obtained from 62 patients with acquired LQTS, the arrhythmia ranged in length from 3 beats (the definition of non-sustained VT) up to 117 beats (Figure 1A), with an average length of 16 ± 8 beats.2 The cycle length (CL) of these episodes ranged from 193 to 364 ms, with an average of 279 ± 47 ms. The VT was frequently preceded by a variable period of bigeminal rhythm due to one or two premature ventricular beats coupled to the prolonged QT segment of the preceding basic beat (Figure 1A and C). This ‘short-long cardiac sequence’ is seen in both acquired and congenital LQTS, and the arrhythmogenic mechanism may be related to increased dispersion of repolarisation (DR).6
Following termination of an episode of fast VT, it is not uncommon to see one or more ectopic beats of variable configuration occurring at much longer CL compared to that of the VT (see beats marked by arrowheads in Figure 2). The change in QRS configuration during VT can take several forms. During a very fast VT, periodic decrease in the amplitude of the entire QRS-T complex is seen with less distinct shifts in QRS axis (Figure 1A). In VTs with slower rates, the classic twisting of the QRS axis from a predominantly positive to a predominantly negative configuration with a variable number of transitional complexes and vice versa is commonly seen as originally described by Dessertenne (Figure 1B).7 Sometimes, a polymorphic QRS configuration is seen without any of the two previously characteristic patterns (as verified in multiple simultaneous leads, Figure 2C, middle recording). Different patterns can be seen in different VT episodes from the same patient (Figure 1C).
QT/T Wave Alternans and Torsade de Pointes
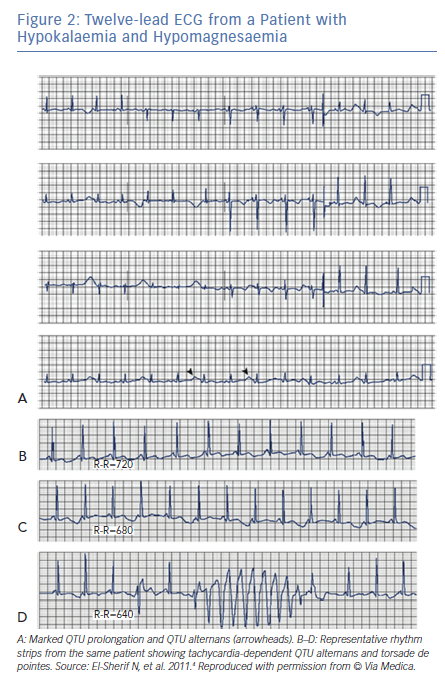
It has long been known that tachycardia-dependent T wave alternans (TWA) occurs in patients with the congenital or acquired form of LQTS and may presage the onset of TdP (Figure 2).2,8
In an analysis of 1,103 LQTS patients with QTc interval >0.44 seconds from the International LQTS Registry, TWA was recorded in 30 patients.8 The frequency of occurrence of TWA was directly proportional to the length of the QTc interval on the enrolment ECG. TWA occurred in one or more occasions during an average 4-year followup in 21% of patients with QTc >0.60 seconds, but in <0.2% of patients with QTc <0.50 seconds. Patients with advanced forms of TWA (those with bidirectional beat-to beat changes in T wave polarity; n=21) were younger, had longer QTc values, had a higher incidence of complex VT, and were more likely to experience a cardiac event (syncope or cardiac arrest) than those with less advanced forms of TWA (those without bidirectional beat-to-beat changes in T wave polarity; n=9).
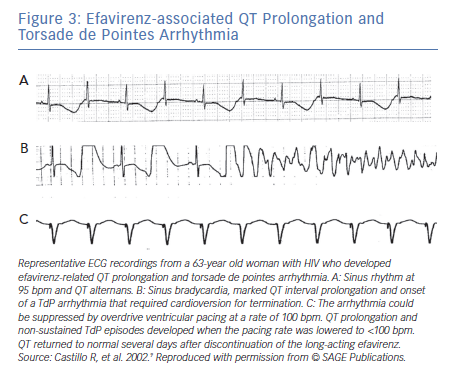
In contrast to TWA in congenital LQTS, the incidence of TWA in acquired LQTS is unknown. It has been reported in acquired LQTS due to hypokalaemia and hypomagnesemia (Figure 3)4 as well as in association with medications that prolong the QT interval (Figure 3).9 Patients with acquired LQTS and TWA are likely to develop TdP (Figures 2 and 3).
Although overt TWA in the ECG is not common, in recent years, digital signal processing techniques have made it possible to detect subtle degrees of TWA.10 This suggests that the phenomenon may be more prevalent than previously recognised and may represent an important marker of vulnerability to VT. A recent report confirms this view, showing that microvolt TWA is far more prevalent in LQTS patients than previously reported and is strongly associated with TdP history.11 Interest in TWA is attributed to the hypothesis that it reflects a greater degree of underlying DR.12
Aetiology of Acquired Long QT Syndrome
The vast majority of acquired LQTS is the result of adverse effects of drugs that interact with the hERG gene, and the IKr. However, although most drugs that cause TdP do so via hERG channel blockade, TdP is not necessarily a potential consequence of all drugs blocking the hERG pathway. Milberg et al.13 compared the TdP induction ability of two hERG-blocking drugs, DL-sotalol and amiodarone. While both drugs can increase the QT interval, the former causes transmural DR and triangulation of the action potential by prolonging phase 3, and triggers both EADs and TdP. However, amiodarone does not usually cause DR, EADs, or TdP, and prolonging phase 2 results in a squared-shaped action potential. While squared-shaped action potentials are considered antiarrhythmic, triangulated action potentials are considered proarrhythmic.14 Several authors have suggested that DR, attributed to preferential prolongation of the APD of M cells, is a preclinical marker of drug-induced proarrhythmia.15 However, the existence, location, and clinical contribution of M cells has been a matter of debate.16
Recent studies have shown that some drugs designated as arrhythmogenic IKr blocker can generate arrhythmias by augmenting INa-L through the PI3K pathway.5 For example, while acute exposure of flecainide to adult mouse cardiomyocyte that lack IKr produced no change in ion currents and action potential duration, extended exposure up to 48 hours of the drug generated up to a 15-fold increase in INa-L and resulted in arrythmogenic EADs. However, not all IKr blockers modulate INa-L, and this diversity of effects, in return, may contribute to the apparent difference in TdP frequency across culprit drugs.5 A major implication of this data has to be that relying on an assay to assesses acute block of IKr may not provide a comprehensive assessment of a candidate drug arrhythmogenic potential.
Antidepressant and antipsychotic drugs modulate the cardiac APD by blocking a variety of cardiac ion channels.17 Some antidepressant and antipsychotic drugs increase the risk of VT and SCD by prolonging the QT interval and inducing TdP arrhythmia. Other antidepressant and antipsychotic drugs increase arrhythmic risk by inducing a Brugada syndrome phenotype. Antipsychotic drugs generally have a higher torsadogenic potential than antidepressants. Based on recent literature, the risk of QT/QTc prolongation with newer non-SSRI antidepressants at therapeutic doses is low.18
Other causes of acquired LQTS include electrolyte abnormalities (hypokalaemia, hypomagnesaemia, and hypocalcaemia [Figure 3]), hypothyroidism, hypothermia, and marked bradycardia, (sinus bradycardia as in Figure 4, or atrioventricular block).19 Any of these factors can cause acquired LQTS or contribute to the risk of drug-induced LQTS. In addition, there is recent evidence of the high prevalence of QTc interval prolongation in patients with anti-SSA/Ro antibodies, as well as autoimmune and inflammatory diseases.20–22
Several recent reports from this laboratory have provided strong evidence for a pathogenic role of autoimmune and inflammatory conditions in the development of QTc prolongation. Anti-Ro antibodies from patients with autoimmune disease were shown to inhibit IKr by directly cross-reacting with the hERG channel, likely at the pore region where homology between 52Ro antigen and hERG channel was demonstrated.23 In addition, an animal model of autoimmune-associated QTc prolongation was established, for the first time, whereby induction of anti-SSA/Ro antibodies by immunisation resulted in QTc prolongation on the surface ECG.23
Conversely, inflammatory channelopathies are related to systemically or locally released inflammatory cytokines (mainly TNF-a, interleukin-1, and interleukin-6) able to directly affect the expression and/or function of several cardiac ion channels, resulting in a decrease of K+ currents (IKr, Ito, or the slow activating components of the delayed K+ current [IKs]) and/or an increase of ICaL.24,25
In another report,26 we tested the hypothesis that IL-6 may cause QT prolongation by suppressing IKr. Electrophysiological and biochemical assays were used to assess the impact of IL-6 on the functional expression of IKr in HEK293 cells and adult guinea-pig ventricular myocytes. In HEK293 cells, IL-6 alone or in combination with the soluble IL-6 receptor (IL-6R), produced a significant depression of IKr peak and tail current densities. Block of IL-6R or Janus kinase (JAK) reversed the inhibitory effects of IL-6 on IKr. In adult guinea-pig ventricular myocytes, IL-6 prolonged APD, which was further prolonged in the presence of IL-6R. Similar to heterologous cells, IL-6 reduced endogenous guinea-pig ERG channel mRNA and protein expression. The data are first to demonstrate that IL-6 inhibition of IKr and the resulting prolongation of APD is mediated via IL-6R and JAK pathway activation and forms the basis for the observed clinical QT interval prolongation. In summary, cardiac or systemic inflammation promotes QTc-interval prolongation via cytokine-mediated effects and this may increase SCD risk.
The ‘Multihit Theory’
A single channelopathy per se is not able in most cases to induce symptoms, and rarely even the related clinical phenotype.25 This is well demonstrated for inherited forms, including LQTS, Brugada syndrome, and catecholamine polymorphic VT, where provocative tests can unmask latent genetic defects. Consistent data are also available for drug-induced, autoimmune and inflammatory/fever-induced channelopathies. Indeed, only a small proportion of the large number of exposed subjects develops drug-induced LQTS-related arrhythmias, despite the resulting channel dysfunction. Similarly, cytokines, and anti-ion channel autoimmune antibodies induce cardiac channelopathy and QTc prolongation. However, inflammatory, and autoimmune-induced phenotypes and arrhythmias occur only in a fraction of the subjects at risk. Such evidence strongly suggests that multiple, often-redundant ion channel mechanisms are implicated in preserving normal AP genesis, thus rendering the clinical phenotype unapparent, despite subtle channel dysfunction. Therefore, more than one single component needs to be impaired for ECG/clinical symptoms to emerge, and the number of required hits will depend on the functional impact of each single offending factor. In a single patient, multiple QT-prolonging factors are concomitantly required to significantly disrupt repolarisation. Accordingly, patients developing marked QTc prolongation and TdP concomitantly present multiple risk factors. In a recent analysis, 18 of 40 consecutive unselected patients with TdP, an average of more than four factors per subject were detectable (electrolyte imbalances, cardiac and extracardiac diseases, drugs, anti-Ro/SSA antibodies, and inflammation), with a high prevalence of acquired channelopathies.24,25
Pharmacogenetics of Acquired Long QT Sydrome
The susceptibility to acquired QT interval prolongation can be influenced by genetic variations.27 This is supported by the fact that the heritability of QT interval duration in the general population (excluding congenital LQTS patients) is estimated to be around 35%.28,29 Further, first-degree relatives of patients with congenital LQTS have a higher risk of drug-induced QT prolongation than non-related individuals.30 In genome-wide association studies (GWAS), a large number of genes associated with QT interval duration has been identified.31 The gene with the strongest signal related to QT interval duration is the nitric oxide synthase 1 adaptor protein gene (NOS1AP), located on chromosome 1 (1q23.3),31,32 which inhibits L-type calcium channel and influences impulse propagation.33 Other findings from GWAS included polymorphisms within genes known to be mutated in congenital LQTS, genes associated with intracellular calcium handling, as well as genes previously not known to influence cardiac repolarisation.34 A recent study that compared 188 patients with drug-induced LQTS and more than 1000 patients with congenital LQTS found disease-causing mutations in 28% of patients with drug-induced LQTS.35 Of interest, under basal conditions, the QTc of drug-induced LQTS patients (453 ± 39 ms) was significantly longer than that of control subjects (406 ± 26 ms).35
It is important to consider pharmacogenetics of drug-induced LQTS as related to both pharmacokinetic and pharmacodynamic properties.27,34 Pharmacokinetics constitute the effect of the body on the drug, which is usually categorised into effect on absorption, distribution, metabolism, and elimination of the drug. Pharmacokinetic genetic susceptibility is mainly characterised by variation in genes encoding drug-metabolising cytochrome P450 or drug transporter like the P-glycoprotein. However, the pharmacodynamics component of genetic susceptibility is mainly characterised by genes known to be associated with QT prolongation in the general population and genes in which the causal mutations of congenital LQTS are located.35
Incidence of Drug-induced Long QT Syndrome
The overall incidence of drug-induced LQTS in a given population is difficult to estimate. One study estimated that between 5% and 7% of reports of VT, VF, or SCD were in fact drug-induced LQTS and TdP.36 European pharmacovigilance centres in Sweden, Germany and Italy have found an annual reporting rate of drug-induced LQTS or TdP of approximately 0.8 to 1.2 per million person-years36 An epidemiological study of drug-induced LQTS in Germany found the reporting rate for symptomatic acquired LQTS to be 2.5% per million person-years for men and 4.0% per million person-years for women, with 60% attributed to drugs.37
QT prolongation is one of the most common reasons for drug withdrawal from the market, despite the fact that these drugs may be beneficial for certain patients and not harmful in every patient.34 Since 1989, 14 clinically important drugs have been removed from the market due to TdP,38 and development of an unknown number has been stopped, due to concerns that these might pose a risk of causing QT prolongation and TdP.38 In the 1990s, the US Food and Drug Administration (FDA) and the European Medicine Agency (EMA) began requiring routine preclinical and clinical testing to determine whether drugs have the potential to cause QT prolongation.39 Today, according to the CredibleMeds website, which has become the standard reference for drug-induced TdP, 38 marketed drugs are recognised for their potential to cause TdP and another 72 to cause QT prolongation.40
In the past decade, hERG channel-mediated cardiac toxicity, manifested as QT interval prolongation, has become a major safety issue in drug development, superseding liver injury as the main cause of drug withdrawals. In vitro electrophysiological testing of the drug’s effects on the function of the hERG channel may be cheaper, faster, and potentially more sensitive than other current surrogates for TdP risk, such as in vivo QT prolongation and action potential prolongation in cardiomyocytes.41

Ethnicity and Gender Differences in Drug-induced Long QT Syndrome
As ethnic differences ultimately reflect genetic variation, it is useful to study ethnicity with regard to susceptibility to drug-induced QT interval prolongation and TdP. However, the role of ethnic differences has not been well established in published studies on drug-induced QT interval prolongation. In 20 QT/QTc studies, only 10% of the total study population was African-American and only 7% was Asian.42 Nevertheless, the frequency of polymorphisms in genes known from congenital LQTS showed varying distribution among ethnic groups.43 African-Americans had the highest risk of prolonged QT interval after acute overdose of QT-prolonging drugs, while Hispanics had the lowest risk compared to all other ethnic groups.44
Females with inherited LQTS demonstrate pronounced gender difference in cardiac repolarisation and arrhythmic risk. Adult women with LQT1 and LQT2 have longer QT intervals, a more pronounced transmural QT dispersion, and a higher risk of TdP and SCD than men.45,46 Interestingly, in female patients with LQT2, the arrhythmogenic risk remains elevated after menopause, suggesting that other gender-related factors besides sex hormones may contribute to gender difference in arrhythmogenesis.47
Gender is a risk factor for adverse drug reactions.48,49 The concept of reduced repolarisation reserve in females compared to males has been used to explain sex differences in arrhythmia risk in acquired LQTS.50 The reduced repolarisation reserve of the female heart is attributed to lower repolarising K+ currents. This difference was thought to be primarily due to testosterone-mediated increase in IKr and IK1 resulting in shorter APD and QTc interval in male hearts. However, in a recent review, the effects of sex hormones go well beyond their modulation of K+ currents.51 The underlying mechanisms can be summarised as follows:52 an estradiol-induced decrease in Ikr as well as increase of ICaL, NCX expression and activity, RyR2 leakiness, Ca2+ transient amplitude, and a1- and b2-adrenoreceptor responsiveness; a testosterone-induced increase in Ikr, Iks, and Ik1, increased SERCA activity, and shortened Ca2+ transient; and a progesterone-induced increase in Iks, increased CERCA expression and activity, and increased ICA-L current sensitivities with reduced Ca2+ oscillations upon sympathetic stimulation. In a recent study, we have shown that modulation of voltage-Ca2+ uncoupling53 may provide one more attractive electrophysiological mechanism for the increased vulnerability of female to drug-induced LQTS.
Acute and Long-term Management of Acquired Long QT Syndrome
The American College of Cardiology (ACC), American Heart Association (AHA), and European Society of Cardiology (ESC) published guidelines for management of ventricular arrhythmias, including drug-induced TdP,54 in 2006 and the key recommendations have been endorsed in a more recent ACC/AHA statement.55
When monitoring for drug-induced prolonged QT interval, a baseline QTc should be obtained. If any one of the following conditions are observed during QT interval monitoring the patient should be admitted to the hospital for telemetry: QTc >500 ms; QTc increase >60 ms above baseline; QT prolongation accompanied by syncope; any evidence of ECG instability, specially TWA, AV block, QRS widening, or ventricular ectopy. The offending drug should be discontinued, electrolyte abnormalities corrected, and a defibrillator placed at bedside.55
Non self-terminating TdP with hemodynamic collapse should obviously be cardioverted with adequate post-cardioversion management. More typical TdP occurs as recurrent self-terminating episodes. In these cases, the first line of management is intravenous administration of magnesium sulphate as a single 2 g (8 mmol) dose over 1–2 minutes followed by a second dose if necessary. Magnesium sulphate is effective in suppressing TdP without reducing the QT interval.56 The mechanism of action may be related to suppression of late calcium influx via L-type calcium current and reduction in the amplitude of EADs.57 If magnesium sulphate fails to suppress TdP, the next step is to increase the heart rate, typically by transvenous pacing. In the interim if necessary, isoproterenol administration could promptly increase the heart rate while waiting for insertion of pacing electrode. Increasing the heart rate is associated with shortening of the QT interval and suppression of TdP. At the same time, the culprit drug should be discontinued and acid-base and electrolytes should be corrected as necessary.
There are novel experimental drugs that can enhance the delayed rectifier conductance52 or activate the cardiac ATP-sensitive potassium channel,58 which may have future value for the treatment of acquired LQTS.
Long-term management of acquired LQTS is important. All patients with previous drug-induced prolongation of the QT interval should be instructed about the importance of subsequent avoidance of QT-prolonging drugs and should have a list of QT-prolonging medications provided by their physicians. In patients with acquired LQTS, the risk of further episodes of TdP is reduced once the culprit drug is removed and other aggravating situations as electrolyte abnormalities or marked bradycardia are corrected. This should result in normalisation of the QTc interval. If it does not, the patient and symptomatic first-degree family members should be considered for genetic testing for the presence of LQTS-associated mutations.55,59 This is especially important for relatives of patients with drug-induced case fatality.
Comprehensive Electrophysiological Mechanisms of Torsade de Pointes
Experimental Models of Long QT Syndrome
Both drug-induced and genetically modified animal models of various species have been generated and utilised to investigate the electrophysiological mechanisms of arrhythmogenesis in LQTS and potential pro- and antiarrhythmic agents. However, due to species differences in features of cardiac electrical function, particularly in repolarisation currents, these models do not completely recapitulate all aspects of the electrophysiology of the human disease. Genetically modified animal models, such as mice and rabbit are commonly used to investigate the arrhythmogenicity of LQTS. Current transgenic LQTS rabbit models have already been instrumental to increasing our understanding of the role of spatial and temporal dispersion of repolarisation to provide an arrhythmogenic substrate, genotype differences in the mechanisms for EAD formation and arrhythmia maintenance, and mechanisms of hormonal modification of arrhythmogenesis.60
In contrast, two dog model of LQTS and TdP have been extensively investigated. One model is the dog with induced complete atrioventricular conduction block (AVB). Complete AVB results within few weeks in hypertrophy and remodelling of the left ventricle associated with prolongation of the QT interval, APD, as well as spatial DR.61 When the animal is challenged with a drug that blocks the IKr, like dofetilide, it results in further prolongation of the QT interval and creation of a drug-induced model of acquired LQTS and TdP.62
The other dog model of LQTS and TdP is the Anthopleurin-A (AP-A) canine surrogate model of LQT3 that was developed in this laboratory.63 The model is created by the neurotoxin anthopleurin-A or ATX2.64 that faithfully reproduces the molecular changes associated with clinical mutations of the Na channel in patients with LQT3.65 Of interest, the experimental surrogate model of LQT3 anticipated the first description of the clinical LQT37 by 7 years.66 Figure 4 is a representative composite of the salient experimental techniques that were utilised to investigate the model and illustrate the correlation between the modulation of a cardiac ion current, its electrophysiological consequence and the final phenotype presentation as LQTS and TdP.
Electrophysiological Mechanisms of the Trigger of Torsade de Pointes in the Long QT Syndrome
There is an almost complete agreement that the initiating one or two beats of TdP are due to EAD-triggered focal activity from the subendocardial Purkinje network.60,66–70 A study by Caref et al. has confirmed beyond reasonable doubt the subendocardial origin of the trigger of TdP.7171 In this study the canine surrogate model of LQT3 was placed on cardiopulmonary bypass and chemical ablation of the endocardial Purkinje network was obtained using Lugol’s iodine following which spontaneous TdP were no longer observed. However, a properly timed premature stimulus-induced reentrant Veterans Administration (VA) based on the underlying marked DR. However, the perpetuation of TdP remains controversial and could be attributed to focal activity, reentrant excitation, or a combination of both mechanisms.
The ionic mechanism(s) that underlie the generation of EADs have been widely investigated. The central hypothesis on the generation of EADs suggests that a spontaneous release of calcium from the sarcoplasmic reticulum would temporarily increase cytosolic calcium concentration with a subsequent sudden activation of the sodium-calcium exchanger inward current. This inward current could ‘re-depolarise’ the sarcolemmal membrane to a potential from which sodium or calcium currents become reactivated, triggering an afterdepolarisation.72 Although this model has been primarily discussed to explain delayed afterdepolarisations, there is also evidence that this mechanism may be the trigger for EADs.73 This hypothesis is also supported by studies that showed that inhibition of the sodium-calcium exchanger suppresses TdP in the intact heart model of LQTS2 and LQTS3.74
Electrophysiological Mechanisms of Perpetuation of Torsade de Pointes
Contrary to the established mechanism of the trigger of TdP, the perpetuation of TdP remains controversial and could be attributed to focal activity, reentrant excitation, or a combination of both mechanisms.75,76 Both reentrant and focal activity are assumed to be non-stationary. For reentrant excitation this could be due to a heterogeneity-induced drift of a reentrant circuit,77 or a meandering reentrant spiral wave78 (Figure 1D). Alternatively, ectopic beats originating from different locations may explain the perpetuation of TdP. The original description of TdP by Dessertenne attributed the pattern to two variable opposing foci (deux foyers)7. In computational modelling as well as experimental observation of the canine chronic AVB model, both multiple competing foci and reentrant excitation could develop depending on heterogeneity of repolarisation in comparison to the surrounding tissue.75 Large heterogeneities can produce ectopic TdP, while smaller heterogeneities will produce reentrant type TdP. The authors of an experimental study in the same model reported that short-lasting episode of TdP had a focal mechanism while long-lasting episodes were maintained by reentrant excitation.76 However, the results were criticised because of the controversial definition of focal versus reentrant excitation.79
It remains an open question as to why and how ectopic beats emerge and compete, and what their relationship is to observe EAD activity. EADs arising from subendocardial Purkinje network conducted to overlying myocardium through Purkinje-muscle junctions (PMJ). Electrotonic interactions across PMJs can modulate APD locally. A recent study has proposed that, dependent on resistive properties across PMJs, large spatial gradient of APD can develop at the endocardium and the transmural plane.80 This may provide a better explanation compared to the concept of mid-myocardial (M) cells with different ionic characteristics.81
One of the problems of sustained fast EAD-induced focal activity is that the short cycle length will be associated with short APD that would suppress further EADs generation unless there is some form of protected islands of prolonged APD with EADs capable of conduction across PMJs to activate the ventricular myocardium. A study that combined computational simulation and experimental observations in isolated myocytes showed that in electrically homogeneous tissue models, chaotic EADs synchronise globally when the tissue is smaller than a critical size. However, when the tissue exceeds the critical size, electronic coupling can no longer globally synchronise EAD, resulting in regions of partial synchronisation that shift in time and space. These regional partially synchronised EADs then form premature ventricular complexes that propagate into recovered tissue without EADs, thus creating ‘shifting’ foci that resemble polymorphic VT.82
Delayed After Depolarisation-triggered Activity Contributes to VA in the Long QT Syndrome
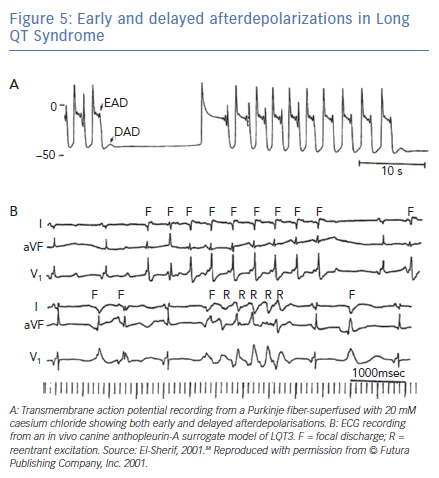
The electrophysiological mechanism of VT in LQTS is somewhat more complex than that described earlier. Figure 5A was obtained from one of the classic reviews of cellular mechanisms of cardiac arrhythmias by Hoffman and Rosen.83 It shows transmembrane AP recording from a canine Purkinje-fiber-superfused with 20-mM caesium chloride (a surrogate experimental model for LQT2). The recording illustrates the classic bradycardia-dependent prolongation of AP duration associated with membrane oscillation on late phase 2/early phase 3 of the repolarisation phase characteristic of EADs. But it also shows that complete repolarisation of the AP is followed by a subthreshold delayed afterdepolarisation (DAD). The latter is simply explained on the basis of increased intracellular Ca2+ associated with the prolonged AP duration triggering a transient inward current. This, almost forgotten, observation strongly suggests that some VT and ectopic beats in LQTS could be secondary to DADs.
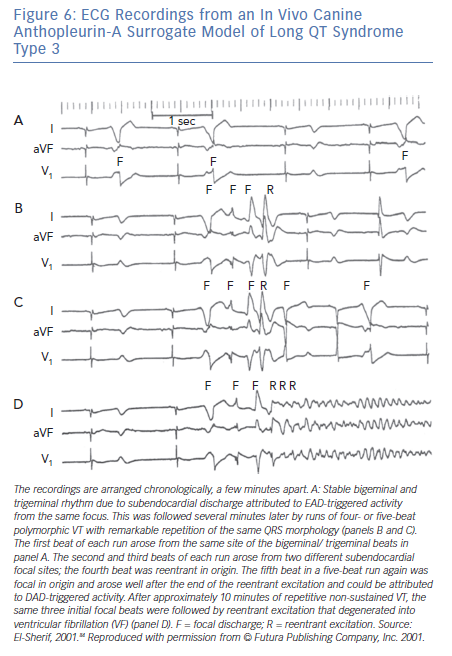
Figure 5B shows a corroboration of this observation from the canine surrogate model of LQT3.84 The top ECG tracing was obtained 10 minutes after infusion of AP-A and shows moderate prolongation of the QT interval and a run of non-sustained monomorphic VT at a rate of 150 bpm. The VT starts with a late coupled beat that is well beyond the end of the QT interval of the preceding sinus beat. Tridimensional mapping of activation showed that the VT arose as a focal discharge (F) from the same subendocardial site. For all practical purposes, the focal discharge could be attributed to DAD-triggered activity.
The bottom ECG tracing was obtained from the same experiment 10 minutes later and shows further prolongation of the QT interval. The ectopic beats labeled F now seem to be coupled to the end of the prolonged QT interval of the preceding sinus beats. The middle of the tracing illustrates a six-beat run of polymorphic VT. Tridimensional mapping shows that the first beat arose from a subendocardial focal site and could be safely attributed to EAD-triggered activity, whereas subsequent beats were due to reentrant excitation in the form of continuously varying scroll waves.
Electrophysiological Mechanisms of Self-terminating versus Non-self-terminating Torsade de Pointes Ventricular Tachyarrhythmia
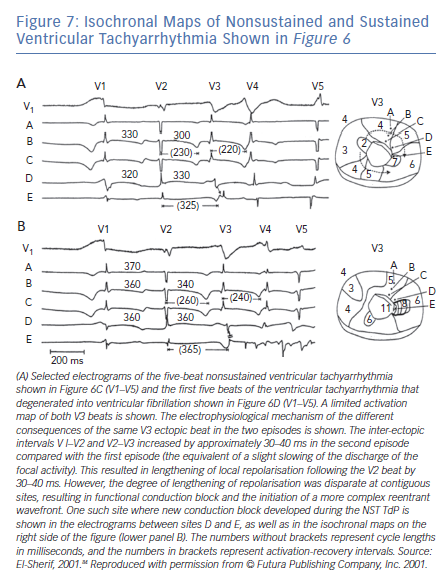
The majority of TdP episodes terminate spontaneously (self-terminating [ST]), (Figure 1). However, a minority can degenerate in VF (non-self-terminating [NST]). The electrophysiological mechanisms of the NST episodes of TdP have never been elucidated. Obviously this is a more important issue than the ‘twisting and turning’ to see if perpetuation of TdP VT is due to focal or reentrant activation.79 Figures 6 and 7 obtained from the canine surrogate model of LQT3 provides one possible electrophysiological mechanism.84 The lesson gained from this example is that subtle changes in underlying spatial DR and conduction characteristics can result in fractionation of activation wavefronts and VF. It also demonstrates clearly the difficulty in predicting which TdP episodes will be ST of NST.
Future Directions
Although congenital LQTS remains the domain of cardiologists, cardiac electrophysiologists and specialised centres, the far more frequently acquired drug-induced LQTS is the domain of all physicians and other members of healthcare teams who are required to make therapeutic decisions. To support better prescribing of medicines, clinical decision support systems to date have issued alerts that warn of potential harm from a prescribing decision.85 However, the impact of these systems has been limited. Moving away from the use of alerts to signal prescribing errors, the concept of ‘medical autopilots’ has been suggested as a preferred approach.86 These programs will monitor the electronic medical record and send signals to guide prescribers toward decisions that result in maximum benefit and minimal risk of TdP.