










Brugada syndrome (BrS), one of the most common causes of sudden cardiac death (SCD) in normal structural heart individuals, is a young entity in modern medicine. BrS was first characterised in 1992 by Brugada et al. as a distinct syndrome with “right bundle branch block, persistent ST elevation in precordial leads V1 to V2–3 and sudden cardiac death”.1 The true prevalence of BrS is not clearly known, with an estimation of 0.05%.2 The prevalence is lower in the Americas and Europe, and higher in Asia, particularly in southeast Asian countries such as Thailand and the Philippines.3
It is hypothesised that BrS accounts for 4–12% of all cases of SCD and 20% of SCD in individuals without structural heart disease.3 The numbers might be underestimated, as suggested by the largest prospective study to date on familial evaluation of sudden arrhythmic death syndrome (SADS).4 With routine application of ajmaline provocation testing and the inclusion of high right precordial leads (RPLs), BrS was shown to be the most prevalent diagnosis (n=85, 28% of families) among inherited cardiac diseases identified in SADS families (n=128, 42% of families).4 The use of high RPLs showed a 16% incremental diagnostic yield of ajmaline testing by diagnosing BrS in an additional 49 families. This study highlights the important role of routine ajmaline testing with high RPLs in improving the yield of diagnostic tests of BrS in SADS families.
The escalating number of publications on this subject in the past 26 years clearly underlines the progress in our understanding and management of BrS. Since the landmark discovery of SCN5A as the first gene linked to the aetiology of BrS, a wealth of knowledge has accumulated in various disciplines including genetics, pathophysiology, clinical manifestations, diagnosis and management, in particular epicardial catheter ablation.5 The complexity of this disease has also been increasingly recognised, with controversies and uncertainties awaiting future studies.
This article provides an update on recent progress in the study of BrS over the past decade.
Progress in Genetic Studies
The first major susceptibility gene reported for BrS is SCN5A.5 Until 2010, almost 300 variations of this gene had been shown to be associated with BrS.6 Despite its role as the major susceptibility gene, SCN5A mutations only account for 11–28% of BrS proband genotypes.6 Other rare gene variations have been reported to be involved in BrS; however, the yield of testing for rare gene variants other than SCN5A has been extremely low.7 Furthermore, differentiation of rare pathogenic variants from rare yet benign variants has been challenging in rare diseases such as BrS. Large-population exome sequencing and in silico tools are likely to be of use in identifying the causative gene variations, although the presence of variants in the general population does not necessarily exclude the pathogenic possibility.8
In 2013, SCN10A was identified by a genome-wide association study as one of the genetic variants that could modulate the susceptibility of BrS (Figure 1A).9 Subsequently, Hu et al. studied 150 unrelated BrS patients and used direct gene sequencing to identify 17 SCN10A mutations in 25 of these patients (16.7%). The identification of SCN10A as a susceptibility gene in this study improved the yield of genotype testing from <35% to >50% of BrS probands.10 However, the monogenic causative role of SCN10A in BrS was questioned by other groups.11,12
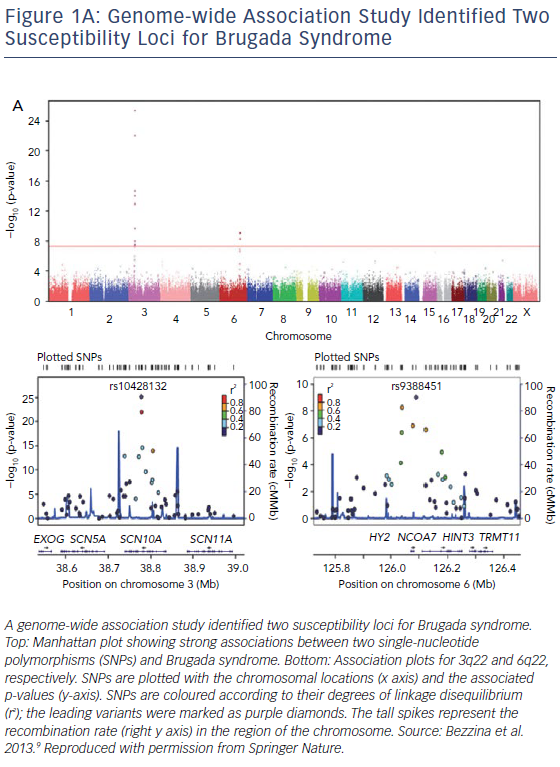
BrS was previously considered a rare disease of single-gene Mendelian inheritance until a genome-wide association study in 2013 demonstrated the strong effect of common genetic variations and polymorphisms on BrS.9 Not only were three common genetic variants (SCN5A, SCN10A and HEY2) identified from the study as modulators for BrS susceptibility, but the risk of BrS also progressively increased in association with the escalating total number of alleles at the three associated loci.9
Models for Pathophysiology
There are three major mechanistic models explaining the electric abnormality in BrS, namely the repolarisation, depolarisation and neural crest models.13–15 Despite their differences, all three models agree that the major region of pathology is the right ventricular outflow tract (RVOT).13–15 Commonly considered as a channelopathy, evidence has revealed structural derangement of the right ventricle in BrS. Using cardiac MRI, BrS patients were found to have right ventricular (RV) motion abnormalities with mildly reduced systolic function and mildly increased RV end systolic volume, compared with normal individuals.16,17 Although late gadolinium enhancement was not detected in cardiac MRI, histological evidence of substantial fibrosis in the RVOT epicardium, corresponding to a low expression of Cx43, was found in the hearts (from autopsy or explanted hearts) of BrS individuals.18 Epicardial and interstitial fibrosis was identified in the slow-conducting RVOT region,18 the ablation of which abolished the BrS ECG pattern.19 A recent study has further shown that RVOT electroanatomical alterations (a low-voltage area) correlate with myocardial inflammation and arrhythmia vulnerability, supporting the hypothesis that BrS is a combination of electrical and structural disease.20
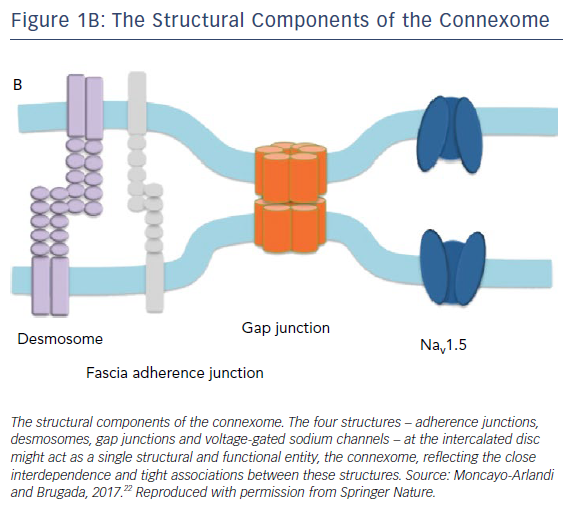
Furthermore, a phenotypic overlap between arrhythmogenic right ventricular cardiomyopathy and BrS has been reported in the literature. RV changes consistent with arrhythmogenic right ventricular cardiomyopathy were observed in patients with clinically diagnosed BrS.21 The concept of the connexome connects the two diseases.22 The connexome is comprised of structures including desmosomes, fascia adherence junctions, gap junctions and voltage-gated sodium channels at the cardiomyocyte intercalated disc (Figure 1B).22
Accumulating evidence has shown that these structures are closely interconnected and interdependent for anchorage and stabilisation. For instance, a mutation in PKP2 – the most important gene responsible for arrhythmogenic right ventricular cardiomyopathy – directly leads to a reduction in Nav1.5 trafficking and activity;23 Cx43 is required for Nav1.5 stability in the intercalated disk membrane.24 Therefore, sodium channel activity could be affected by the disruption of any connexome components.22
Update on Clinical Diagnosis
A recent change in the BrS phenotype at presentation compared with earlier years (prior to 2003) has been noted.25 There is a decreased number of patients presenting with aborted SCD, spontaneous type 1 ECG pattern and arrhythmia inducibility during electrophysiology study (EPS), whereas the prevalence of syncope remains stable.25 This shift to a milder clinical profile is likely owing to better identification and thus improved diagnosis of BrS.
The diagnostic criteria have been updated in the 2013 consensus statement by the Heart Rhythm Society (HRS), the European Heart Rhythm Association (EHRA) and the Asia Pacific Heart Rhythm Society (APHRS).26 Contrary to the prior 2005 HRS/EHRA criteria, the 2013 HRS/EHRA/APHRS consensus statement has listed several differences. Firstly, the ECG pattern criteria for diagnosing BrS are different. The 2005 criteria required ST elevation in ≥2 RPLs (V1–V3) in a standard position (the 4th intercostal space) for the diagnosis of type 1 BrS.2 In the 2013 consensus statement, a type 1 Brugada ECG pattern in ≥1 RPL (V1–V2), whether in a standard or a higher position (the second and third intercostal space),26 was promoted, which increases the sensitivity of diagnosis. Secondly, the 2005 criteria required at least one of the clinical presentations (documented ventricular tachycardia [VT] or VF, syncope, nocturnal agonal respiration family history of SCD or type 1 ECG, or ventricular arrhythmia inducibility in EPS) for diagnosis.2 However, clinical presentations are no longer essential for diagnosis in the new consensus statement.26 BrS could be definitively diagnosed based on ECG pattern and after the exclusion of other differential diagnoses (e.g. atypical right bundle branch block, early repolarisation, MI, arrhythmogenic right ventricular cardiomyopathy).26 Thirdly, the 2013 consensus statement has weakened the importance of differentiating between spontaneous type 2 and type 3 BrS ECG patterns, and highlights the importance of the type 1 ECG pattern, whether it is spontaneous or drug induced.26 The 2013 consensus statement recommendation for diagnosing BrS was subsequently adopted by the 2015 European Society of Cardiology guidelines for managing ventricular arrhythmias and preventing SCD.27
Advances in Management
Risk Stratification for ICD
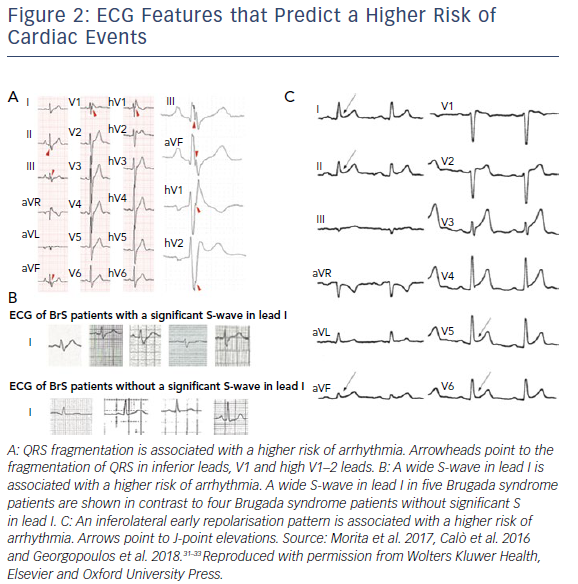
Symptoms (including syncope and aborted SCD) and a spontaneous type 1 BrS ECG pattern are known to carry a significantly higher risk of ventricular arrhythmia and SCD in BrS patients.28–30 Aside from a spontaneous type 1 ECG pattern, other ECG features such as QRS fractionation (HR 4.94), a wide S-wave in lead I (HR 39.1) and inferolateral early repolarisation (HR 4.87) have also been shown to portend a high risk of future ventricular arrhythmia in BrS patients (Figure 2).30–33
To date, ICD placement is the most accepted therapy for preventing SCD in high-risk BrS patients. On the other hand, long-term complications of ICD, including inappropriate shocks, infection, injury and device malfunction, can significantly increase patient health burden and decrease quality of life.34 Current guidelines recommend ICD placement in individuals with aborted SCD (class Ia), syncope (judged likely to be secondary to ventricular arrhythmia) and a spontaneous type 1 ECG pattern (class IIa), and ventricular arrhythmia inducibility during programmed stimulation study (class IIb).26
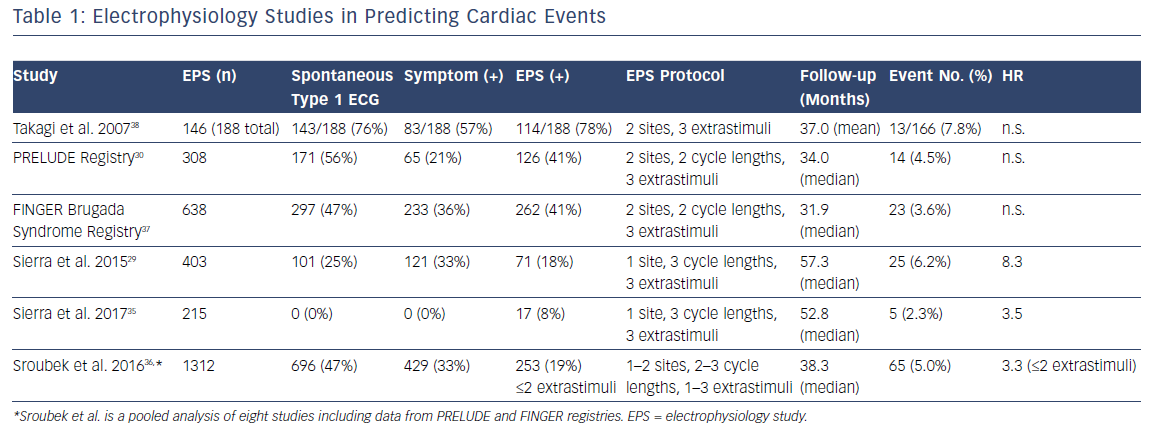
The role of EPS in the risk stratification of BrS patients has been debated for years, yet it still remains controversial owing to inconsistent results among different studies29,30,35–38 and meta-analyses.39,40 The reason for different observations in various studies is likely multifactorial, including patient populations (e.g. the percentage of patients with symptoms at presentation, spontaneous versus drug-induced type 1 ECG pattern), different protocols for programmed stimulations (e.g. stimulation sites and numbers of extrastimuli) in different centres, as well as follow-up durations (Table 1). Sroubek et al. performed a pooled analysis using individual-level data from eight studies (1,312 BrS patients) and found that arrhythmia inducibility was associated with an increased risk of cardiac events (HR 2.66, multivariate analysis).36
Notably, although several early studies applied two stimulation sites (RV apex and RVOT), a less aggressive strategy involving only RV apex is currently recommended to increase the specificity of the test.41 Furthermore, the study by Sroubek et al. showed that up to two extrastimuli was associated with increased risk of future arrhythmic events, while up to three extrastimuli reduced the test’s specificity.36
With a milder current clinical profile of BrS patients compared with earlier years, ventricular arrhythmia inducibility during EPS might lose its predicting power owing to significantly lower pre-test probability and the need for a longer follow-up duration.25 On the other hand, a negative EPS study does not eliminate the future risk of ventricular arrhythmia,except potentially in asymptomatic patients with drug-induced type 1 ECG.35,36 A recent study from the Survey on Arrhythmic Events in Brugada Syndrome (SABRUS) – a multicentre international survey that included BrS patients with arrhythmia events – showed that among the BrS patients who exhibited arrhythmic events after prophylactic ICD, 25% did not meet the criteria for class II indications at the time of ICD implantation.42 This group either had negative EPS, or EPS was not performed. These findings argue for a better stratification strategy among BrS patients in the future.
A multiparametric approach has been attempted aiming at better risk stratification.43–45 An escalating accumulation of multiple high-risk factors (e.g. family history of sudden death, positive EPS, syncope and type 1 ECG pattern) predicts a progressively increased risk of future SCD or ventricular arrhythmic events.43,44 A score model recently proposed by Sieira et al. reported a high predictive performance of 0.82; however, further validation is required.45 This model includes spontaneous type 1 ECG, early family history of SCD, EPS inducibility, syncope, sinus node dysfunction and SCD, and gives a higher score for symptoms and EPS inducibility (Figure 3).
Medications
Quinidine – a class I anti-arrhythmic medication and an Ito current inhibitor – is an established medication used to prevent and terminate ventricular arrhythmia, electrical storm and frequent electric ICD shocks in BrS patients.46,47 Anguera et al. conducted a multicentre study in Spain and found that 29 of 820 BrS patients (3.5%) with ICD were treated with quinidine for electrical storm or frequent ICD shocks.47 During a follow-up of 60 ± 41 months, 19 patients (66%) remained free of ICD shocks. Quinidine treatment decreased the total shocks from 203 to 41, with the median number of shocks per patient decreased from 6 (interquartile range [IQR] 4–9) to 0 (IQR 0–2.5).40

Others have explored quinidine as a potential therapy for long-term SCD prevention, especially among asymptomatic patients with arrhythmia inducibility.48–50 Belhassen et al. studied 96 BrS patients, in which 66 patients (10 patients with previous aborted SCD, 20 with syncope and 40 asymptomatic) were induced with VF using an aggressive stimulation protocol (two sites and up to three extrastimuli).49 Quinidine was able to prevent VF induction in 52 of the 58 (89.6%) patients tested. During a median follow-up of 113 months, the overall annual arrhythmic events were 1%. However, the caveats of this study included an aggressive stimulation protocol and low long-term medication adherence (60%), due to side-effects.49
Low tolerance and medication adherence caused by the side-effects of quinidine have been a concern. It has been shown that the risk of arrhythmic events in quinidine recipients was associated with medication interruption.40,49 Side-effects are reported to be as high as 36–38% of patients administered a daily dose of quinidine bisulfate (QBS) 1,500 mg or hydroquinidine (HQC) 900 mg.39,49 Medication discontinuation was reported at 14–30% due to medication intolerance.41,49 Diarrhoea is the most common adverse effect (9–18%), and other side-effects include thrombocytopenia (6.6–13.6%), and less commonly allergic reactions (<5%), oesophagitis (<5%), side node dysfunction (<5%) and lupus-like reactions (<5%).39,49 QTc prolongation was often observed (the change by percentage was reported from <10% to 15.8%); however, torsades de pointes was not reported.47–50 A lower dosage (daily HQC dose 600 mg) appears to be better tolerated.50,51 Further lower dosages (QBS or HQC ≤600 mg) have been reported to reduce side-effects while maintaining efficacy;52 however, concerns exist regarding the reduced suppression of VF inducibility and possible compromised anti-arrhythmic effects at doses lower than 600 mg.51
Despite great efficacy in treating BrS, quinidine is currently an endangered species due to the shrinking pharmaceutical market for quinidine in the era of newer anti-arrhythmic medications for common cardiac arrhythmias. AstraZeneca was the main quinidine manufacturer, but stopped production in 2006. According to a survey of physicians in 131 countries, limited access to quinidine was reported in 76% of countries (including Thailand and the Philippines, where BrS has a high prevalence).53 Even in the US, where quinidine is still produced, it is not readily available in many healthcare facilities.
The use of isoproterenol infusion is a class IIb recommendation for electrical storm in BrS patients.26 Other oral medications suggested as long-term alternatives to quinidine have been explored. Cilostazol, bepridil and denopamine, alone or combined, have been reported to prevent VF storm in BrS patients in clinical cases after initial stabilisation with isoproterenol.54–56 Cilostazol, an inhibitor of phosphodiesterase III, increases inward the calcium current and suppresses the Ito current by increasing heart rate. Case reports have demonstrated its efficacy in suppressing recurrent VF and VT.57,58 However, its failure to suppress VF storm in BrS patients has also been reported.59,60 Bepridil is a multichannel blocker that inhibits L- and T-type calcium channels, as well as all potassium channels. Aizawa et al. reported the efficacy of bepridil efficacy in suppressing recurrent VF in five BrS patients (a total of 19 shocks were given in a year before bepridil and two shocks while on bepridil).56 The combination of bepridil and cilostazol has been proposed for its possible synthetic effects in VF prevention and the attenuation of cilostazol-induced palpitations.55 This regimen was shown to effectively suppress recurrent VF and ICD shocks in five BrS patients (13 ICD shocks in total in an accumulated period of 55 months before medication, and 0 shocks in an accumulated 272 months while on medication).55 Despite the promising results, the numbers of patients in these studies are low, with relatively short follow-up periods. Direct comparison with quinidine has also not been studied. Future studies are needed to further address these issues.
Radiofrequency Ablation
Radiofrequency ablation (RFA) has arisen as a promising therapeutic option for BrS in the past decade. Studies have demonstrated that in addition to abnormally low voltage, prolonged duration and fractionated electrograms, histological/molecular changes including inflammation, fibrosis and low expression of Cx43 have also been found at the anterior RVOT epicardium.18–20 These findings have established the foundation of ablating the substrate as a feasible treatment for BrS. The current consensus recommendation considers RFA as a reasonable therapy for BrS patients with arrhythmic storms or repeated appropriate ICD shocks (Class IIb).26 With the progress made in recent years, there is hope that RFA might even offer a ‘cure’ for selected patients.61
RFA Approaches: Endocardial versus Epicardial
Despite early attempts for ablation beginning with endocardial approaches (endocardial ablation of arrhythmogenic premature ventricular contraction (PVC) and endocardial mapping with ablation of late-activation zone),62,63 later studies have shown that most of the electrophysiological substrate locates at the RVOT epicardium,19,64–66 and thus epicardial ablation has become a more accepted approach due to its improved efficacy in eliminating arrhythmogenic substrates.
A recent meta-analysis including 11 case series and 11 case reports (total number of patients: 233) has provided a systemic overview on the evidence of RFA in BrS.67 A comparison was made between the following ablation strategies: epicardial mapping with substrate ablation (n=180), endocardial-only mapping with ablation (n=17), VF-triggering PVC ablation (n=5) and mixed approaches (n=30). Elimination of type 1 Brugada ECG pattern was achieved in 98.3% of the epicardial approach groups versus 34.8% in the endocardial approach.67 The success rates in preventing VT/VF were 96.7%, 70.6% and 80.0% in epicardial, endocardial and triggering PVC ablation strategies, respectively.67
Since the landmark study in 2011 by Nademanee et al. – the first report of successful epicardial ablation in nine patients with BrS and frequent VF episodes – recent years have seen advances in substrate mapping and ablation, as well as end point tests of successful ablation (Table 2).19
Substrate Mapping and the Use of Sodium Channel Blockers
Endocardial and epicardial electroanatomic mapping of RV is widely performed in order to identify the substrate in BrS patients.19,36,64–66,68,69 Notably, in patients undergoing both epicardial and endocardial mapping, no endocardial substrate was identified in 93% of cases.67 Areas with abnormal electrograms (low voltage, fractionation, prolonged duration or late potentials) are marked as substrate for ablation. The definition of abnormal electrograms has been slightly different in various studies: the cutoff of low voltage ranges between 1.0 mV and 1.5 mV, fractionation is defined as less than two versus three components, and prolonged duration is defined as >80 ms versus 200 ms (80 ms in most of the studies) (Table 2). Notably, assessment of a low-voltage area could vary depending on tissue contact, pericardial fat and pericardial fluid during flushing.61 Avoiding the sole use of low-voltage criteria is recommended to increase the accuracy of substrate mapping.61
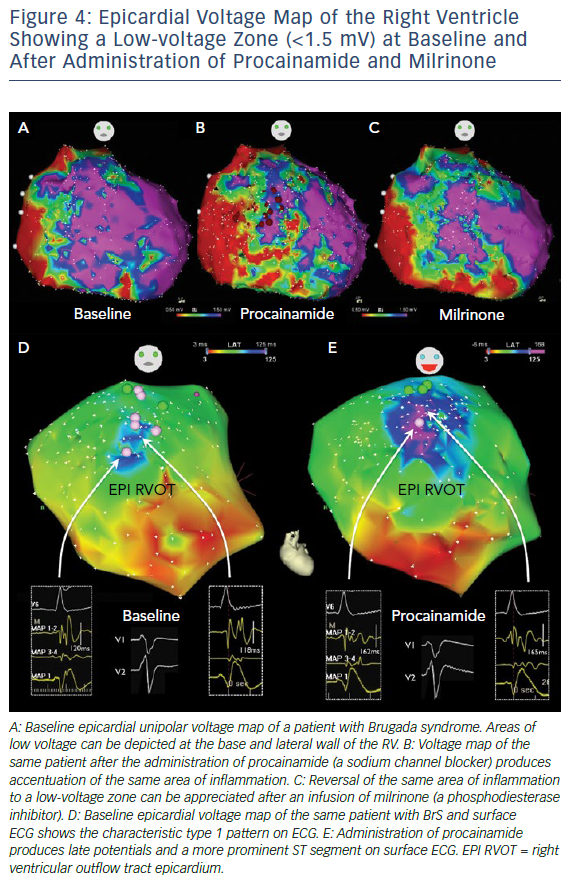
Sodium channel blockers have been used to increase the sensitivity of substrate identification (Figures 4 and 5). In 2015 Brugada et al. reported the successful identification and epicardial ablation of substrate with the additional use of flecainide, which increased the abnormal electrogram area from 17.6 cm2 to 27.3 cm2.66 The increased sensitivity of substrate mapping with a sodium channel blocker was echoed in other ensuing studies.19,64 A report including 135 BrS patients (with spontaneous or induced VT/VF) further demonstrated the efficacy of the epicardial mapping and substrate-based ablation approach in preventing future VT/VF.64 In this study, Pappone et al. showed that the areas of substrate were larger in symptomatic BrS patients, in contrast to asymptomatic patients at diagnosis (4.6 cm2 versus 8.0 cm2 in patients with induced VT/VF versus patients with aborted SCD).64 Ajmaline infusion expanded the substrate area (the median area increased from 4.6 cm2 to 15.7 cm2 in patients with induced VT/VF, and from 8.0 cm2 to 20.0 cm2 in patients with SCD at presentation), corresponding with an increase in type 1 ST elevation on ECG.
More recently, Pappone et al. demonstrated that after ajmaline administration, patients with no prior arrhythmia inducibility developed inducibility without any significant difference in substrate characteristics.70 Moreover, substrate size was found to be the only predictor of inducibility (OR 4.51, 95% CI [2.51–8.09], p<0.001), with a size of 4 cm2 more commonly observed in patients with inducible arrhythmias (area under the curve 0.98, p<0.001). Substrate ablation was associated with ECG normalisation and not arrhythmia re-inducibility.70 These observed correlations further suggest that improving the sensitivity of substrate identification is the key to improving ablation efficacy.
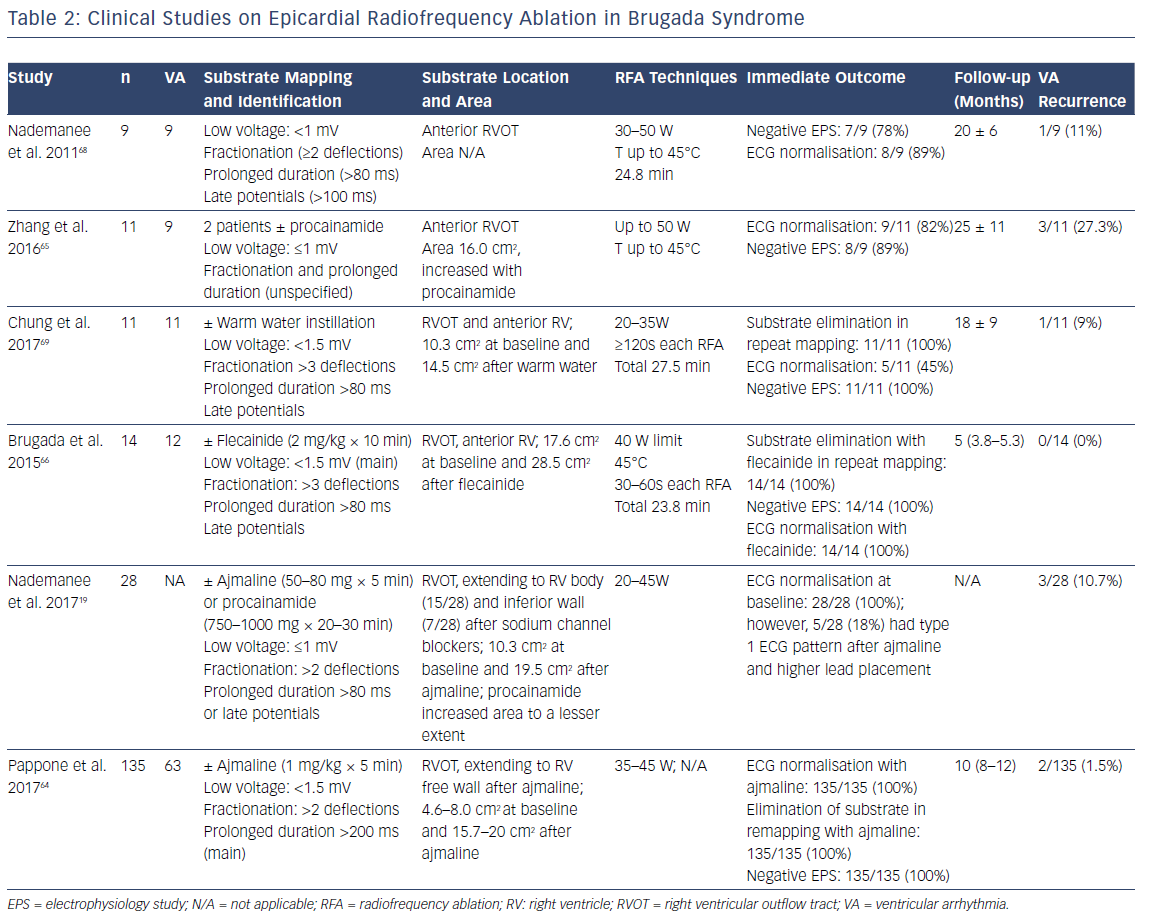
Similar to diagnosing the drug-induced type 1 BrS ECG pattern, the sodium channel blockers ajmaline (1 mg/kg × 5 min), flecainide (2 mg/kg × 10 min) and procainamide (750–1,000 mg for 20–30 min) have been utilised in different studies (Table 2). Procainamide appears to increase the substrate to a lesser extent than ajmaline.19 There is no direct comparison between ajmaline and flecainide. However, in diagnosing the drug-induced type 1 ECG pattern, a higher sensitivity of ajmaline (1 mg/kg × 5 min) over flecainide (2 mg/kg × 5 min) was reported.71
Attenuation of Brugada Syndrome Phenotype by General Anaesthesia
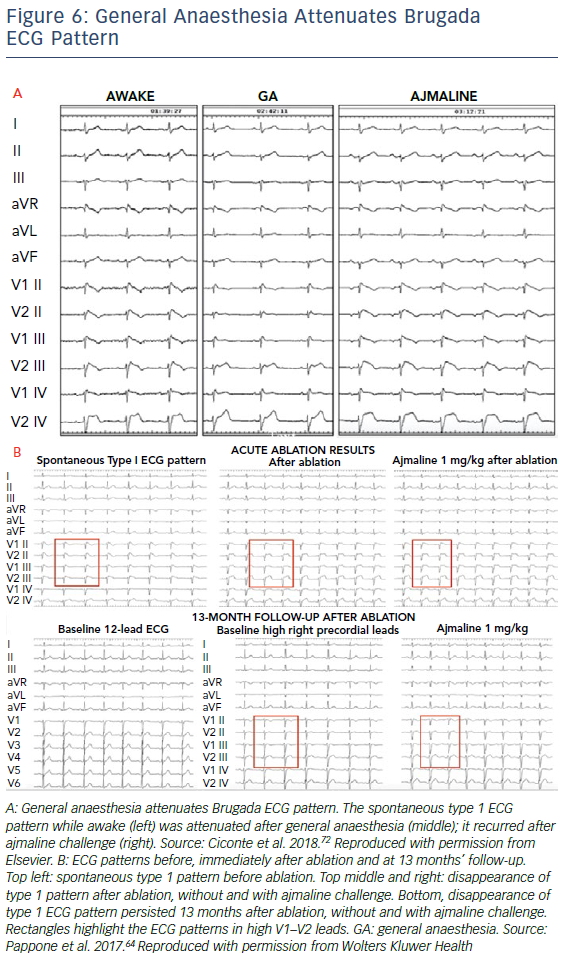
An observation of the effect of general anaesthesia on BrS phenotype during RFA in BrS patients was recently reported.65 General anaesthesia was induced by propofol bolus and maintained by sevofluorane. After the induction of anaesthesia, ST elevation (0.32 ± 0.01 mV versus 0.19 ± 0.02 mV, p<0.001) and J-wave amplitude (0.47 ± 0.02 mV versus 0.31 ± 0.03 mV, p<0.001) significantly reduced. The ECG pattern was reversed to a nondiagnostic pattern in 28 out of 36 (77.8%) patients (Figure 6A). However, the arrhythmogenic substrate area during anaesthesia was still significantly enlarged after administration of ajmaline (3.6 ± 0.5 cm2 versus 20.3 ± 0.8 cm2).65 This study has demonstrated a significant clinical implication in assessing the substrate area during RFA under anaesthesia.
Efficacy of the Radiofrequency Ablation Procedure
Technical advances in ablating catheters might have improved the efficacy of substrate elimination.19 A contact force of at least 5 g is recommended to effectively ablate lesions with a radiofrequency power of 20–45 W.19 The electrogram voltage amplitude drastically decreases during RFA. The reduction of voltage to <0.5 mV (dense scar tissue), with the disappearance of the mid and late components of fractionated potentials, indicates the elimination of the arrhythmogenic substrate (Figure 5).19,64
In studies conducted thus far, various tests have been utilised as evidence of successful ablation, including normalisation of type 1 ECG pattern (Figure 6B), abolishment of VT/VF inducibility and elimination of substrate in remapping (Figure 5). Persistent or recurrent J-ST elevation has been shown to be associated with recurrence of VT/VF after RFA.67 In a 2017 report by Nademanee et al., the authors recommended eliminating all substrate areas with abnormal low-voltage and fractionated electrograms, confirmed by repeat substrate mapping, rather than using VT/VF inducibility or ECG normalisation as the end points for epicardial ablation.19

Ongoing Clinical Trials
Despite various studies reporting on the success of epicardial ablation with low recurrence of VT/VF, the sample sizes in most of these studies are small. Additionally, the lack of control groups, different protocols employed (including substrate mapping and different sodium channel blockers used) and varying follow-up durations in each study make it difficult to draw clear conclusions regarding the efficacy of RFA for treating BrS. A multicenter study (Bangladesh Risk of Acute Vascular Events [BRAVE] study, ClinicalTrials.gov identifier NCT02704416) has commenced and is currently in the recruiting phase to test the efficacy of epicardial ablation in BrS patients to prevent VT/VF episodes. An estimated 184 patients will be randomised to two arms: continued ICD therapy (control arm) or epicardial ablation plus continued ICD (intervention arm). This study is expected to be completed in 2021.
Conclusion
This review summarises progress in the understanding and management of BrS in recent years. Not only has new knowledge been acquired in the genetics and molecular mechanisms of BrS, but recent years have also seen progress made in risk stratification as well as the development of promising new therapies, including epicardial ablation for BrS. Future studies are needed to further clarify the pathogenesis of this complex disease and to guide clinical practice, including genetic testing, risk stratification and selection of therapies.
Clinical Perspective
- The complexity of Brugada syndrome (BrS) continues to evolve, and therefore understanding the mechanisms of this disease is imperative to provide better alternatives for treatment.
- Early detection and risk stratification are areas of major importance in the treatment of BrS.
- Radiofrequency ablation of RVOT substrate has arisen as a promising treatment modality for BrS, although larger scale and long-term follow-up studies are required to further determine
its merit.