









Worldwide obesity has reached pandemic proportions with more than 1.9 billion adults classed as overweight in 2016, of which 650 million were obese.1 Since it is a major modifiable risk factor for so many cardiovascular diseases, it is no surprise there has been an exponential increase in cases of AF coinciding with the rise in obesity. While there were an estimated 8.8 million cases of AF in 2010 in Europe alone, by 2060, this is estimated to rise to 17.9 million.2 Moreover, obesity is now the second biggest attributable risk factor for AF after hypertension. Together with overweight, it accounted for 17.9% of all AF cases in the Atherosclerosis Risk in Communities (ARIC) study.3 While AF risk appears to follow a linear pattern with increase in BMI, the pathophysiological basis of the obesity–AF relationship is complex and multifactorial.4 Indeed, various epidemiological studies have demonstrated an apparent paradox with regard to outcomes in AF patients; overweight and mildly obese patients with AF appear to have an overall better prognosis in terms of all-cause mortality compared with lean patients with AF.5 This is consistent with other cardiovascular diseases.
In this review, we will begin with a detailed discussion of the epidemiological links between obesity and AF highlighting the benefits and relative limitations of using BMI and other anthropometric markers in assessing adiposity (obesity is typically defined as BMI >30 kg/m2). We will outline general mechanisms contributing to AF and place obesity in this context, focusing on the pathophysiological mechanisms that underpin the obesity–AF relationship with emphasis on recent insights derived from studies on adipose tissue biology. Finally, we discuss novel therapeutic targets and the importance of a holistic approach to dealing with this burgeoning public health issue.
Epidemiology of the Obesity–AF Relationship
Early associations between AF and obesity were observed in patients undergoing cardiac surgery, with high BMI being reported in numerous studies as a major risk factor for post-operative AF.6–8 Various studies including the Framingham Heart Study and a meta-analysis has indicated that a rise in BMI parallels a marked increase in AF risk (Table 1).9,10 The Women’s Health Study found that for every 1 kg/m2 increase in BMI, there was a 4.7% increase in risk of developing AF.4 In a cohort of 47,589 patients prospectively followed up for a mean of 5.7 years in the Danish Diet, Cancer and Health Study, BMI independently correlated with increased AF risk regardless of gender.11 In a recently published cohort of 67,238 patients derived from a database of healthcare claims in the US, obesity was associated with new onset AF independent of age, diabetes, hypertension and gender.12
Notably, BMI has even been associated with progression of AF from paroxysmal to persistent in a two large community cohort studies.13,14 The mean age of patients in the Olmsted County study by Tsang et al. was 71, suggesting that this relationship may not necessarily be applicable to a younger cohort of patients.13 However, a subsequent retrospective Danish registry study of 271,203 women who had given birth found that while the overall AF incidence remained low in this young population, obesity remained a significant independent predictor for AF.15 Indeed, the generalisability of the obesity-induced AF risk remains true across geographical and racial boundaries. Berkovitch et al. demonstrated in a large Israeli cohort of 18,290 men and women that overweight and obesity were associated with a higher AF risk and that weight reduction corresponded to a reduced risk of developing AF.16 They reported a 7% reduction in AF incidence for every 1 kg/m2 drop in BMI. Much of the obesity–AF risk is often attributed to other coexisting cardiovascular risk factors given obesity is associated with conditions such as diabetes and hypertension. A Korean study attempted to account for this by reviewing a cohort of so-called metabolically healthy obese (MHO) patients; those with a high BMI but who were free of other morbidity.17 They retrospectively reviewed nearly 400,000 patients and found the MHO cohort also had an increased risk of AF development; on multivariate analysis they had a hazard ratio of 1.3 compared with their healthy non-obese counterparts. This would suggest that the obesity–AF relationship appears to stand beyond conventional risk factors.
The Obesity Paradox and AF
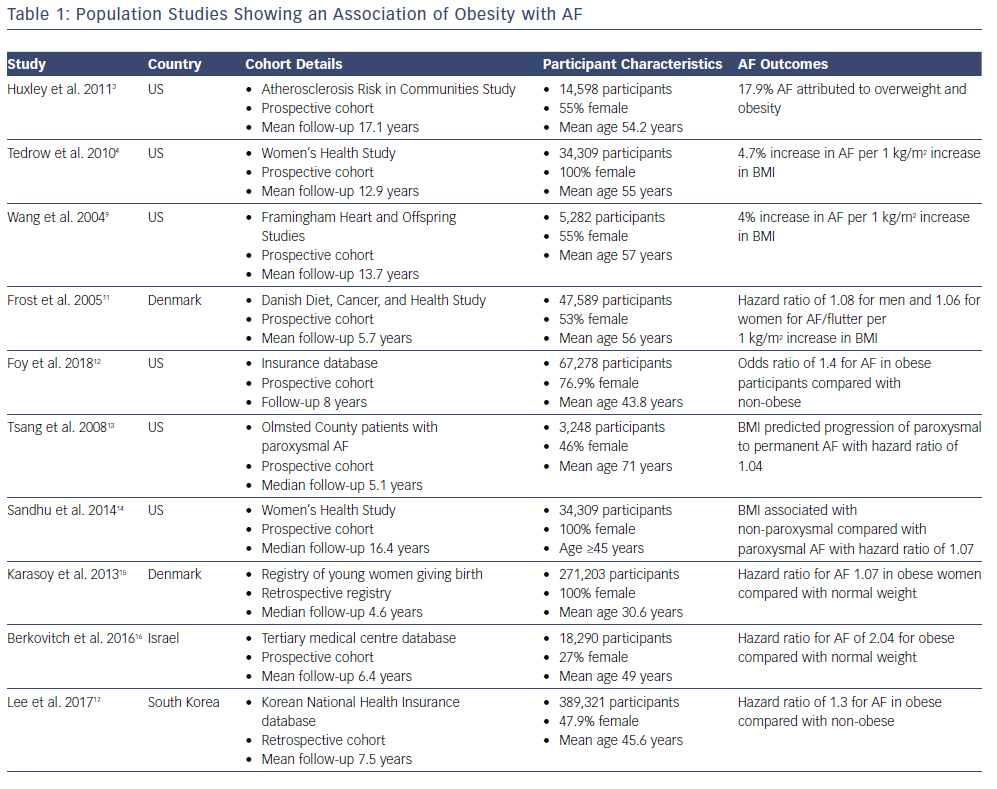
While there is a consistently and extensively reported relationship between increasing BMI and AF risk, progression and recurrence, a counter-intuitive opposite effect is observed regarding mortality. Known as the obesity paradox, overweight (BMI 25–30 kg/m2) and mildly obese (BMI 30–35 kg/m2) people appear to have lower all-cause mortality in long-term follow-up studies.18–20 Indeed, this phenomenon stands true for many cardiovascular diseases.5 A meta-analysis pooling nine studies involving 49,364 participants found that underweight (BMI <18.5 kg/m2) Asian patients with AF were at increased risk of embolic events, such as stroke and systemic embolism, as well as cardiovascular and all-cause death.21 Additionally, they found that in all AF patients, overweight and obesity were not associated with these outcomes.
In order to dissect the potential reasons for this paradox, it is important to consider the characteristics of the patients included in these studies. The authors allude to various confounding factors that could account for this phenomenon. First, a greater proportion of patients with AF in cohort studies tend to be overweight or obese (78% in the Pandey study) while those patients who have normal BMI tend to be significantly older (in the Sandhu et al. cohort, obese patients were significantly younger).18,19 Age is particularly relevant given it is a major predictor of all-cause mortality in AF.22 Second, there appears to be a greater use of rhythm control strategies and anticoagulation in patients with high BMI, potentially to account for a greater proportion of persistent AF in groups with a higher BMI.18 Third, patients with high BMI tend to have higher blood pressures facilitating greater use of appropriate cardiac medications.23 Fourth, patients with an apparently normal BMI may have other medical conditions which lead to a relatively catabolic or pro-inflammatory state and that increased BMI provides a metabolic reserve in this case. Fifth, cardiorespiratory fitness is being increasingly recognised as a major factor in reducing AF.
Qureshi et al. demonstrated in a large multi-racial cohort of 64,561 adults that for every one metabolic equivalent achieved during treadmill testing, there was a 7% lower risk of incident AF even when accounting for potential confounders.24 In the CARDIO-FIT study, Sanders et al. showed in 308 patients with AF that for every two metabolic equivalents gained following their risk factor management and tailored exercise programme, there was a twofold improvement in arrhythmia-free survival.25 This was accompanied by reduced AF burden and an improvement in symptoms. More recently, Malmo et al. found that after a 12-week interval training programme for patients with paroxysmal AF, AF burden, as measured by implantable loop recorders, reduced by 50% compared with controls.26 Additionally, the authors observed a trend towards fewer cardioversions and hospital admissions alongside significant symptomatic improvements.
The BMI Effect
There are relative pros and cons of using BMI as a measure of adiposity. BMI is the most widely used marker of obesity with obvious advantages in terms of ease of obtaining height and weight data on patients. However, it fails to capture body fat distribution or take into account body composition.27,28 For instance, athletes with a high muscle mass and a higher weight may fall into an overweight category despite having a relatively low body fat composition. On the other hand, central abdominal obesity is associated with deleterious outcomes.29 Various other anthropometric markers, such as waist circumference and waist-to-hip ratio, may better indicate the distribution of adiposity and better reflect risk. Indeed, lean body mass was recently reported in a Danish registry cohort as the predominant anthropometric risk factor for AF with other markers not showing an association with AF when adjusted for lean body mass.30 In a cohort of 130,473 UK Biobank participants without any smoking-related disease or weight loss, no significant difference in mortality was seen between normal and overweight groups (stratified by BMI).29 However, in each group, a higher waist-to-hip ratio reflecting central adiposity was associated with excess mortality risk.
Taken together, there are numerous potential causes that could account for this apparent obesity paradox seen with AF and other cardiovascular conditions (Figure 1). Furthermore, additional unaccounted confounding factors and selection bias, such as a genetic predisposition to AF, also need to be considered. Certainly, given the improvements observed in AF outcomes with risk factor management and weight loss, the apparent obesity paradox should not deter from an aggressive risk factor optimisation strategy to manage AF.
The Pathophysiological Context of the Obesity–AF Relationship
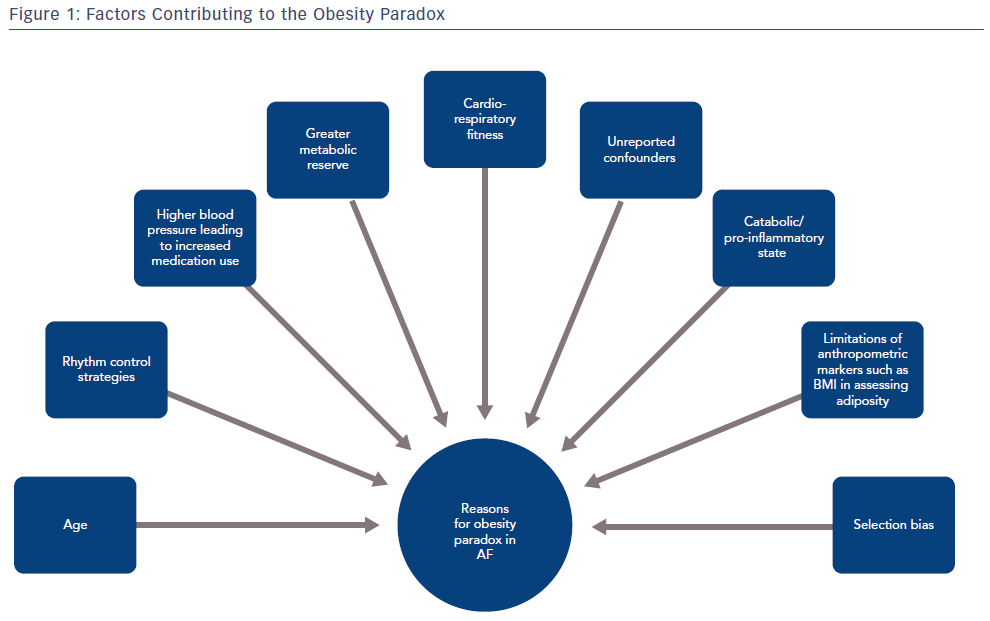
Obesity sits alongside a number of conventional cardiovascular risk factors that often accompany it and increase AF risk, such as diabetes, hypertension, metabolic syndrome and ischaemic heart disease. These factors will promote structural atrial remodeling, fibrosis, wave break, micro reentry and AF, even in the absence of obesity. Importantly, some of the mechanistic pathways that lead to AF in these conditions are shared with obesity–AF pathophysiology. For instance, hypertension induces haemodynamic changes such as increases in left ventricular filling pressures, stiffness and diastolic dysfunction that are also seen in obese states.31 Moreover, activation of the renin-angiotensin-aldosterone system typically observed in hypertensive patients, is similarly seen in obesity.32 Adipocytes have been documented to directly produce aldosterone, and aldosterone antagonists have been noted to be particularly effective in patients with heart failure with reduced ejection fraction with associated abdominal obesity.33–35 In the case of diabetes, persistent hyperglycaemia is associated with the development of advanced glycation end-products which can infiltrate the myocardium provoking fibrosis and hypertrophy, in turn distorting the architecture of the heart to make it more conducive to AF genesis.36,37 While this can be independent of obesity, the fibrotic mediators, such as the transforming growth factor beta (TGF-beta) superfamily, are common to both pathways.36 Similarly, diastolic dysfunction is common in the diabetic heart and is similar to that seen in obesity.
Factors such as sleep apnoea/obesity hypoventilation syndrome (often known together as sleep-related breathing disorders) are emergent players to consider. In a study looking at patients being evaluated for bariatric surgery, sleep-related breathing disorders were present in 88% of patients.38 Moreover, the Sleep Heart Health Study demonstrated that patients with sleep-related breathing disorders were at fourfold increased risk of AF and threefold increased risk of non-sustained ventricular tachycardia following adjustment for several confounders.39 We will discuss the mechanistic basis for this in the following section.
Obesity-Related Haemodynamic Changes
Obesity is associated with a host of haemodynamic changes that alter cardiac structure and physiology to make it more conducive to AF development and maintenance. Adiposity is associated with an increase in total blood volume with this typically resulting in increased cardiac output. With little change in heart rate, the rise in cardiac output is predominantly due to an increase in stroke volume. Unfortunately, a sustained rise in cardiac output is associated with left ventricular enlargement and eccentric or concentric hypertrophy.40 With raised left ventricular filling pressures, diastolic dysfunction occurs and over time with persistently raised filling pressures and left ventricular hypertrophy, systolic impairment may also occur.41 In addition to this, increased BMI is associated with left atrial enlargement with raised left atrial pressures and volumes.42 A consequent increase in pulmonary capillary pressures result. With obesity commonly associated with sleep-related breathing disorders, the cycle of repeated hypoxia, acidosis and arousal from sleep can alter autonomic tone, increasing the risk of abnormal cardiac impulse formation, as well as increasing pulmonary arterial pressures which in turn can lead to right ventricular hypertrophy and failure.39 Left unchecked, biventricular hypertrophy and impairment can result with associated distortion in left atrial architecture and haemodynamics providing the ideal substrate for AF genesis and persistence.
Epicardial Fat, Adipose Tissue Biology and AF
Given adipose tissue distribution appears to be a key factor in determining cardiovascular risk, it is no surprise that the role of distinct adipose tissue depots is of intense interest.43 With more sophisticated cross-sectional imaging modalities such as CT and MRI being increasingly used for cardiac imaging, the visceral fat layer directly overlying the myocardium – epicardial adipose tissue (EAT) – can be directly imaged. Of note, pericardial fat is often referred to interchangeably with EAT in the literature but strictly speaking paracardial fat refers to the layer of fat external to the parietal pericardium and pericardial fat is the combination of both EAT and paracardial fat.44
EAT covers 80% of the heart’s surface and up to 20% of the heart’s weight, predominantly overlying the coronaries, atrioventricular and interventricular grooves but also spanning the atria and ventricles.45,46 There are no fascial boundaries between EAT and the myocardium and they share the same blood supply, the coronary vessels, facilitating direct paracrine and vasocrine effects on the heart.47 EAT’s physiological roles include thermoregulation, a source of energy and mechano-protection.47,48 A seminal post-mortem study assessing adiposity of the heart in 1933 showed that 98% of obese patients had excessive epicardial fat.49 Cross-sectional imaging data has indicated EAT volume correlates with increased AF risk with left atrial EAT volume a particular predictor of AF.50–52 Worse outcomes following AF ablation and also post-operative AF in the cardiac surgical setting have been reported.51,53 A meta-analysis confirming this association between EAT and AF has shown an overall odds ratio of 2.61 per each standard deviation increase in EAT volume with an even higher odds ratio for persistent AF.54
Role of Inflammation
EAT is a highly active visceral tissue producing a host of different pro- and anti-inflammatory adipocytokines, metabolic and growth factors that can directly diffuse into the myocardium.45,48 Various lines of evidence suggest local inflammation is a key mediator in AF. In a study looking at the immune cell profile of patients undergoing valve surgery with either no history of AF or those with persistent AF, the number of CD45+ cells (a pan-leukocyte marker) was significantly higher in the atrial myocardium of AF patients.55 This was corroborated in a study by Smorodinova et al. demonstrating that not only were there higher CD45+ cell populations in the AF cohort but specifically CD3+ T lymphocytes and CD68+ cells (likely corresponding to dendritic cells) were significantly higher in the AF cohort.56 In coronary artery bypass surgery patients, where AF is a common post-operative complication, high levels of pro-inflammatory cytokines including tumour necrosis factor-alpha (TNF-alpha), interleukin-1beta (IL-1beta) and IL-6 were observed in the EAT compared with subcutaneous adipose tissue samples.57 AF patients undergoing valve surgery were found to have higher levels of right atrial nuclear factor-kappa beta (NF-kappa beta), which is a key regulator of the immune response. Additionally, TNF-alpha and IL-6 levels were higher and increased fibrosis and severe lympho-monocyte infiltration were seen.58 Clearly, local inflammation mediated by a locally driven immune cell and cytokine response appears to play a key role in AF pathophysiology and is likely to underpin the EAT-AF relationship.
Fibrosis and Lipotoxicity
Alongside inflammation, fibrosis is recognised as a central factor in the development of an arrhythmogenic substrate.41 Pro-inflammatory cytokines and growth factors such as activin A and matrix metalloproteinases are likely to mediate a fibrotic effect on the atrial myocardium via paracrine pathways.59,60 In a study by Venteclef et al, only the EAT secretome – and not the subcutaneous secretome – applied to an atrial preparation induced myocardial fibrosis with the myofibroblasts able to produce extracellular matrix components.59 This fibrotic effect was attributed to activin A which reproduced the significant fibrotic effect when applied to the atrial preparation with upregulation of pro-fibrotic genes. The fibrotic effect was negated by application of an activin-A antibody.59 Activin-A is a member of the TGF-beta superfamily and induced expression of TGF-beta1 and beta2 in the same study.59 TGF-beta1 has been found to cause selective atrial fibrosis and increase vulnerability to AF when overexpressed in a transgenic mouse model.60 Fibrosis plays a major role in creating the electrical heterogeneity, regions of local conduction block, changes in atrial refractoriness and the formation of reentrant circuits that ultimately form the substrate for AF.61,62
While inflammation and fibrosis play central roles in the obesity–AF cascade, direct fatty infiltration of the myocardium in obesity is also likely to play a role in inducing deleterious structural and electrical changes in the atria.63 Mahajan et al. used an ovine model of obesity with sheep fed a high-fat diet for a period of 72 weeks.63 Extensive imaging and electrophysiological analysis showed that the sheep displayed a higher AF burden with enlarged atria and EAT fatty infiltration of the posterior left atrial wall. This was accompanied by significant atrial fibrosis, reductions in conduction velocity in both atrial chambers and reduced posterior left atrial endocardial voltages. These insights into adiposity and AF must be put into context of the study. It used a laboratory animal model of obesity with sheep fed a high calorie and high fat diet that may not necessarily reflect the model of obesity seen worldwide, which is caused by Western diet and sedentary lifestyle. The authors acknowledge that it is not possible to draw a causal link between fatty infiltration and AF from this study.63
Direct Electrophysiological Effects of the Secretome
In addition to the inflammatory and fibrotic mediators that may underpin the obesity–AF relationship, visceral fat and its secretome has been reported to have direct electrophysiological effects in left atrial (LA) cardiomyocyte co-culture experiments in rabbits.64 Lin et al. demonstrated that epicardial, abdominal and retrosternal adipocytes all prolong the LA action potential, while epicardial adipocytes also significantly altered the resting membrane potential.64 The late sodium current, L-type calcium channel current and transient outward current were all increased in the co-cultured cells while delayed rectifier potassium currents were smaller.64 The authors also noted greater isoprenaline-induced delay after depolarisations in the co-cultured cells.64 Taken together, these changes in action potential would all promote arrhythmogenicity in left atrial cardiomyocytes and illustrate the extensive direct modulatory effects of visceral fat.
Autonomic Nervous System in Obesity
Autonomic dysfunction secondary to obesity with concomitant sleep apnoea has been shown to trigger AF in animal models.65 Moreover, EAT contains the ganglionated plexi that are thought to be key mediators of autonomic modulation of the heart.66 In a canine model by Po et al., parasympathomimetics were injected into EAT resulting in bradycardia followed by premature depolarisations and subsequent spontaneous AF.67 The site of firing was thought to arise from both the pulmonary vein and non-pulmonary vein sites. Further evidence of the role of ganglionated plexi in AF comes from ablation of the plexi as part of AF ablation procedures, which tends to diminish AF inducibility.68
A recent study looked at a cohort of paroxysmal AF patients undergoing coronary artery bypass surgery where botulinum toxin targeting the ganglionated plexi was injected into the EAT at the time of operation. The investigators found that there was a significant reduction (7% versus 30%) in the percentage of patients having postoperative AF in the botulinum toxin group at 30 days with a sustained difference seen at 1-year follow-up.69,70 The mechanism for this observed effect is likely to involve targeting the deleterious role of the autonomic nervous system on AF inducibility. Botulinum toxin inhibits the exocytic release of acetylcholine from pre-synaptic vesicles affecting parasympathetic cholinergic neurotransmission.71 When injected into the epicardial fat pads which play host to ganglionated plexi, which harbours predominantly parasympathetic neurons, it is likely to cause temporary suppression of cholinergic firing. AF vulnerability is linked to the duration of the atrial effective refractory period (ERP) and the dispersion of the ERP.72 Vagal activation shortens the atrial ERP and increases dispersion. A canine study has demonstrated increased ERP dispersion and increased AF vulnerability after toxin was injected into the epicardial fat pads.72 With regard to the longer-term suppression of AF reported in the study, the study authors refer to autonomic hyperactivity and AF forming a cycle with autonomic activity triggering AF and then AF further enhancing autonomic activity.73 The authors allude to ‘breaking this cycle’ thus preventing the atrial remodelling associated with prolonged periods of AF and reducing the vulnerability to episodes of AF in the longer term.70
The obesity–AF relationship is complex with multiple insults, such as inflammation, fibrosis, lipotoxicity and autonomic dysregulation, combined with haemodynamic and mechanical changes forming the substrate and trigger for AF. With accompanying obesity-related comorbidities such as diabetes, hypertension, sleep-related breathing disorders and ischaemic heart disease included in this picture, an optimal milieu for AF maintenance is created. Figure 2 summarises these mechanisms.
Management of AF in Obese Patients
Drug Therapy
The effect of obesity on AF risk extends to altering aspects of management in AF patients. One of the pillars of AF management is anticoagulation to minimise thromboembolic complications associated with the condition. In a recent study reviewing warfarin dosing in patients stratified by BMI, participants with a high BMI, particularly more than 40 kg/m2 had significantly higher warfarin requirements.74 A higher weekly dose of warfarin may have implications for time to discharge if the drug is commenced in hospital or in maintaining time in therapeutic range. It would seem that the use of direct oral anticoagulants (DOACs) including dabigatran, apixaban, rivaroxaban and edoxaban for thromboembolism prophylaxis would address this issue. However, there is a paucity of large-scale clinical trial data or pharmacokinetic analyses in patients of high BMI with most of the data gleaned from subgroup analyses.75
Guidance from the International Society on Thrombosis and Haemostasis suggests avoidance of DOACs in morbidly obese patients (BMI >40 kg/m2) or with a weight of >120 kg, due to limited clinical data.76 Yet this approach would exclude a large number of patients who may benefit from DOACs. Indeed, in a study of healthy volunteers with a weight of more than 120 kg who were taking rivaroxaban, the differences in factor Xa inhibition were 10% lower compared with those of normal weight.77 Kaplan et al. recently evaluated obese patients including those with a BMI >40 kg/m2undergoing direct current cardioversion (DCCV) for AF or atrial flutter on DOACs and warfarin and found there was a very low incidence of stroke with none seen in the BMI >40 kg/m2 cohort at 30 days.78 While the patient cohort group consisted of only 761 patients, this study would suggest DOACs appear to be safe in a cohort who have a relatively elevated risk for stroke in the first month post-DCCV.
AF Procedures
A second pillar of AF management is rhythm control, with one of the most commonly performed procedures for patients in AF being DCCV. This facilitates prompt evaluation of symptomatology and, in the longer term, the assessment of changes in cardiac dimensions and function when in sinus rhythm. In turn, this should guide ongoing management. Patients with a higher body weight have been found to have a lower success rate with cardioversion.79 This is likely to be due to a lower energy being delivered to the heart in patients with a higher body weight with higher energies in a subsequent study being associated with a greater likelihood of successful cardioversion in obese patients.80 Higher energies would achieve increased local atrial current densities to depolarise both atria simultaneously and re-establish sinus rhythm. A recent randomised study sought to identify additional strategies alongside higher energy delivery that would enhance the success rate of the procedure.80 The authors found that the use of paddles (rather than adhesive patches), manual pressure applied by two operators with a gloved hand when patches are used, as well as escalation of energies up to 360 J, would enhance the likelihood of successful cardioversion in obese patients.
Catheter ablation is a principal rhythm control tool with various studies demonstrating that a higher BMI corresponds to a higher AF recurrence risk.81–84 While complication rates generally do not differ in the studies, greater radiation exposure was noted in one study.83 Winkle et al. noted that minor complications were significantly higher in the morbidly obese group (BMI >40 kg/m2), although no differences in major complications were observed.81 The authors allude to the role of weight loss strategies to optimise the success of AF ablation.
Novel Therapeutic Opportunities
Epicardial Adipose Tissue Biology
Having extensively reviewed the critical local role of EAT in mediating the obesity–AF relationship, it is no surprise that it is an emerging therapeutic target. Arresting the inflammatory cascade in EAT by targeting specific adipocytokines using antibody approaches would help to reduce this major component of AF genesis and maintenance. Similarly, antibodies to pro-fibrotic agents such as TGF-beta, ideally targeted to the adipose tissue for instance with nanoparticle technology, would help to improve selectivity of delivery and minimise off-target effects, improving safety.85
Agents that reduce EAT volume have also been investigated. In a cohort of patients with diabetes, Iacobellis et al. demonstrated a near 40% reduction in EAT (as measured by echocardiography) in the group given liraglutide, a glucagon-like peptide 1 (GLP-1) agonist in addition to metformin.86 There was no double-blinding or reporting of cardiovascular outcomes but liraglutide and GLP-1 analogues could be a new area of therapeutic investigation. Indeed, GLP-1 receptors are present in adipose tissue and appear to regulate adipogenesis providing some mechanistic explanation of the phenomenon observed.87 The sodium-glucose co-transporter 2 inhibitor dapaglifozin has been shown to produce loss in body weight and concomitant reduction in EAT volumes as well as reduction in systemic TNF-alpha levels.88 Moreover, dapiglifozin-treated EAT explants demonstrated lower levels of chemokines compared with untreated explants.89 Further work to assess AF outcomes in patients with diabetes treated with these agents would help guide the use of these medications in reducing AF risk among this group.
Role of Imaging
EAT has been shown to be an independent risk factor in AF incidence and progression across a range of different settings. Cross-sectional imaging approaches such as CT and MRI may help to form novel algorithms to predict AF risk and progression. This would be particularly relevant to AF ablation. Standards and guidelines with regards to cut-off EAT volumes to predict AF risk would need to be well-validated in large cohorts for them to have widespread clinical utility. One study looking at 283 patients with AF looked at echocardiography-derived EAT thicknesses and suggested using cut-off EAT thicknesses values of 6 mm for paroxysmal AF and 6.9 mm for persistent AF.90 While echocardiography is widely available and inexpensive, there are significant limitations with regards to reproducibility and lack of volumetric data. Moreover, quantification of EAT sub-depots, for instance around the left atrium, is not possible. Antonopoulos et al. used adipose tissue CT attenuation to create the fat attenuation index, a new metric to study adipose tissue.91 The fat attenuation index reflects the degree of adipocyte differentiation and lipid accumulation in fat depots.91 The metric correlates well with coronary inflammation, reliably separating stable and unstable lesions in acute coronary syndromes.91 In the case of AF, a similar technique may help to identify EAT that is particularly inflamed and hence more likely to be pathogenic.
Risk Factor Management
Sanders et al. have extensively contributed to the evidence base for a comprehensive risk factor management approach to AF.92–6 This approach is akin to the secondary prevention approach used for patients with ischaemic heart disease. There have been trials that look at the role of weight loss alongside risk factor optimization, such as good diabetic control, achieving hypertension targets, lipid-lowering strategies, smoking cessation and alcohol reduction, as well as improving cardio-respiratory fitness.25,92–6 This has produced significant benefits in reducing the burden of AF. Moreover, reverse cardiac remodelling and even a regression from persistent to paroxysmal or no AF has been demonstrated.92,95 Notably, a dose-dependent relationship between weight loss and freedom from AF was observed.93 The value of a physician-led service was highlighted, given 84% patients who achieved >10% weight loss were in the physician-led clinic.93 The cost-effectiveness of such intense risk factor management was evaluated by the same group who found that with less medication needed, fewer hospitalisations, emergency attendances and procedures, the cost-saving benefit amounted to US$12,094. This corresponded to an increase of 0.193 quality-adjusted life years.96
Dietary intervention and bariatric surgery are other avenues to facilitate weight loss. Data remains largely speculative with regard to dietary approaches to reducing AF risk while bariatric surgery can achieve significant sustained weight loss and concomitant large reductions in AF risk.97 In the Swedish Obese Subjects cohort study following-up 2,000 patients undergoing bariatric surgery, a 29% reduction in AF risk compared with controls was observed at follow-up of 19 years.98 Differences in AF rates were only seen between the groups after 5 years, highlighting the importance of sustained weight loss and the temporal delay in altering the AF substrate.
Primary prevention of AF in the obese at-risk cohort would be the next step in helping to control the obesity–AF epidemic. For instance, a combination of imaging to calculated threshold volumes of visceral fat (such as EAT) to guide risk stratification and using drugs such as GLP-1 analogues and sodium-glucose co-transporter 2 inhibitors in people with diabetes and obesity.
Conclusion
The seemingly inexorable rise of AF cases worldwide is paralleled by the obesity epidemic. Their associated morbidity and mortality combined with huge financial costs mark out the obesity–AF relationship as one of global health importance. The epidemiological associations suggest that obesity increases the risk of AF incidence, progression and recurrence. The numerous pathophysiological mechanisms highlight the complexity of the relationship but also illustrate the potential for new therapeutic opportunities such as targeting the inflammatory-fibrotic cascade. Moreover, a comprehensive risk factor management approach appears to be a realistic, cost-effective and efficacious solution to addressing this problem. Long-term studies will confirm whether such strategies yield improvements in hard outcomes such as mortality.
Clinical Perspective
- Obesity is a key driver in the exponential rise of AF cases worldwide.
- High BMI is a major predictor of AF incidence and progression.
- Epicardial adipose tissue (EAT) is the visceral fat layer directly surrounding the myocardium and it plays a critical role in obesity–AF pathophysiology. EAT’s secretome of inflammatory mediators and pro-fibrotic molecules contribute to the substrate of AF, and EAT harbours the ganglionated plexi that can trigger AF.
- Cross-sectional imaging of visceral fat together with antibody-based therapeutic agents targeting the inflammatory-fibrotic cascade may prove to be novel tools in AF management.
- A comprehensive risk factor management programme is both a clinically and cost-effective strategy to manage AF.