










AF is the most common sustained cardiac rhythm disturbance.1 Worldwide, there is an estimated 33.5 million people with AF as of 2010.2 While there are multiple estimates, the yearly incidence in the US is expected to grow, for example, from 1.2 million cases in 2010 to 2.6 million cases in 2030 and upwards of 6–12 million cases by 2050.1,3 It is associated with increased morbidity and mortality4 and diminished quality of life.5 AF is associated with structural heart disease, but it may also occur in a structurally normal heart.
The clinical evaluation of patients with AF predominantly focuses on upstream risk factors, such as: hypertension, diabetes, heart failure (reduced or preserved ejection fraction), valvular heart disease, and other cardiopulmonary pathology such as pulmonary embolism.6,7 Despite being the site of origin for this arrhythmia, the atria are not typically evaluated in any systematic manner except for atrial size and/or volume, typically evaluated on echocardiography. Furthermore, atrial size and left atrial (LA) appendage (LAA) morphology have not been incorporated into clinical decisions such as starting anticoagulation in patients with AF.7 For example, the guideline-recommended and widely used CHA2DS2-VASc score for the evaluation of stroke risk does not include atrial size or any measure of atrial pathophysiology.8 Ample data have shown that the atria are diseased in many patients with AF.9,10 In this paper, we will explore the pathophysiology of atrial myopathy, the clinical evaluation of atrial fibrosis, the relationship of atrial fibrosis with thromboembolic events, and finally potential treatment approaches that might affect this atrial myopathy.
Pathophysiology of Atrial Myopathy
The development of atrial myopathy is complex with multiple factors contributing to its development. Here, we will discuss the development of atrial fibrosis, the role of obesity, the interaction of autonomic dysfunction and comorbidities on atrial myopathy, and the link between AF and atrial myopathy.
Development of Fibrosis
Fibrosis is a common pathway to injury and failure in multiple organs. Four steps in the fibrogenic cascade have been identified: initiation of the body’s response to the initial injury; activation of effector cells; elaboration of extracellular matrix (ECM); and finally the development of fibrosis with organ failure from this matrix.11 Tissue damage initially leads to both a regenerative phase where tissue is replaced by normal tissue cells with no permanent damage, and a fibrosis stage in which normal parenchyma is replaced with fibroconnective tissue.12 This second phase is regulated by a complex pro-inflammatory cytokine- and cell-mediated system in which fibroblasts and myofibroblasts synthesise fibrotic tissue.11,12
This process of fibrosis after injury is well characterised, for example, in the development of ischemic ventricular cardiomyopathy after MI. The resultant necrosis from an initial MI triggers the innate immune pathway activation of nuclear factor (NF)-kB.13 In addition, growth factors such as transforming growth factor-beta 1 (TGF-beta1), endothelin 1, and angiotensin II will cause the proliferation of fibroblasts and myofibroblasts. These processes drive the inflammatory response and lead to ECM deposition and fibrosis.11,14 Two types of fibrosis occur in the heart: reactive fibrosis, which occurs in the perivascular space and is similar to the fibrosis seen in other tissues; and replacement fibrosis, which occurs at the site of prior myocyte loss. This fibrotic remodelling of the heart leads to the systolic and diastolic dysfunction seen in heart failure.15,16
There is a similar role for fibrosis in the development of atrial myopathy. The TGF-beta1/Smad signalling pathway has been identified as a key pathway in the development of atrial fibrosis. TGF-beta1 is a member of the cytokine family responsible for tissue repair and the generation of fibrosis. Smad is an additional family of proteins that can both attenuate and inhibit TGF-beta1 to help regulate this pathway.17 In the rapid pacing animal model for AF, activated TGF-beta1 phosphorylates Smad, to form complexes that translocate into the nucleus of myofibroblasts to upregulate the production of fibrin with resultant atrial fibrosis. In addition, overexpression of TGF-beta1 decreases the production of Smad7, which is responsible for the inhibitory feedback loop of the TGF-beta1/Smad pathway.17,18 The renin–angiotensin–aldosterone system (RAAS) has also been implicated in the development of atrial fibrosis. Studies of transgenic mice with overexpression of angiotensin-converting enzyme (ACE) have noted marked atrial dilation and fibrosis.19,20 Furthermore, the rapid pacing of atrial myocytes has been shown to cause the expression of microRNA that alter fibroblasts to increase ECM production.21 As discussed later in this paper, these various proteins integral to the development of atrial fibrosis can serve as both serum biomarkers and therapeutic targets.
Role of Obesity
Obesity in general has been linked to a higher incidence of the development of AF.22–24 More specifically, epicardial adipose tissue (EAT), which refers to the layer of adipose tissue overlying the myocardium,25 has the potential to directly affect the atrial myocardium via the release of adipokines that promote inflammation and fibrosis.26,27 EAT has been shown to secrete activin A (a member of the TGF-beta family) that promotes the development of atrial fibrosis via a paracrine effect.28 EAT can also serve as a source of progenitor cells that can differentiate into the myofibroblasts that are responsible for the creation of ECM and fibrosis.29 A link between EAT and AF can be seen clinically. Patients with AF have been found to have higher levels of EAT compared with controls;30 furthermore, subjects with chronic AF are more likely to have a higher volume of EAT than those with paroxysmal AF.30 On multivariate analysis there was an independent association between EAT and LA volume even when controlling for BMI.30 Obese patients have increased low-voltage areas (13.9% versus 3.4%, p<0.001) and low-voltage areas correspond to the location of EAT on MRI.31 Furthermore, EAT has been shown to be a predictor of the success of AF ablation and the risk of recurrence.32–35 EAT has also been shown to be an independent risk factor for AF-associated stroke.36
Other Components of Atrial Myopathy
The occurrence of atrial fibrosis in AF is clear. While this may play a critical part in the pathogenesis of AF via conduction slowing and the facilitation of re-entry, it is important to acknowledge that there are also other components of the atrial myopathy that may also be implicated. For example, there are autonomic innervation changes that occur after rapid pacing. Arora et al. showed that in the canine model, after rapid pacing, there is an increase in both sympathetic and parasympathetic fibres in the posterior LA and pulmonary veins. These autonomic changes cause regional shortening of the atrial effective refractory period, creating a substrate for re-entry and AF.37–39 These changes in innervation of the cardiac autonomic ganglia are present in AF patients and are an integral part of the atrial myopathy and have been suggested as a target for ablation.40
The systemic diseases incorporated in the CHA2DS2-VASc score, such as hypertension, heart failure, and diabetes, have been associated with increased risk for AF and may also play a role in the development of atrial myopathy.10 These comorbidities have been associated with increased LA pressure in AF patients.41 Hypertension is associated with abnormalities in the RAAS with high circulating levels of angiotensin II, ACE, and aldosterone. In addition to the fibrotic mechanism previously discussed, these components contribute to atrial myopathy by affecting ion channel structure and function and cause pro-inflammatory effects on the LA.42 Furthermore, left ventricular hypertrophy as the result of longstanding hypertension has been associated with both LA thrombus and increased risk of stroke.43–46 Heart failure (both preserved and reduced ejection fraction) causes increased LA pressure, increased LA size, and upregulation of RAAS.46 Patients with diabetes have increased angiotensin II levels, TGF-beta signalling, adipose tissue, and systemic inflammation.47 Patients with abnormal glucose metabolism have larger atrial sizes, lower atrial voltage, and higher AF recurrence after ablation.48 It is clear that multiple pathophysiologic pathways may be implicated in the formation of the atrial myopathy depending on the upstream initiators.
Patients with atrial myopathy have been found to have endothelial dysfunction leading to increased hypercoagulability.49 Rapid atrial pacing of pigs has been shown to increase the levels of the endothelial nitric oxide synthase inhibitor asymmetric dimethylarginine (ADMA).50 Furthermore, in patients with persistent AF, there are higher levels of ADMA.50 ADMA leads to endothelial dysfunction and increases the risk of stroke.51 Additionally, von Willebrand factor levels have been associated with both AF and the risk of ischemic stroke.52,53 These prothrombotic factors are accentuated in patients with comorbid hypertension, heart failure, and diabetes by increased oxidative stress.40
AF and Atrial Myopathy
Cardiac fibrosis is a key anatomic mediator creating the electrophysiologic substrate for arrhythmias.11,54–56 This has been illustrated in the animal model in which selective creation of atrial fibrosis via overexpression of TGF-beta1 has been shown to lead to increased inducibility of AF.57,58 Likewise, fibrosis creates a heterogenous milieu that slows intercellular conduction.39,59–61 There is an alteration in cell–cell connections, gap junctions, and anisotropy that contributes to re-entrant pathways.40 In patients undergoing catheter ablation for AF, atrial tissue conduction velocities were decreased in fibrotic areas defined by low voltage.62 These areas of slowed conduction velocity were predictive of sites in which ablation either terminated AF or slowed the AF cycle length by at least 30 ms (sensitivity 72.0% with 95% CI [50.6–87.9%] and specificity 78.1% with 95% CI [71.3–80.9%]).62 Furthermore, atrial fibrosis allows for the formation of multiple re-entry circuits that are thought to perpetuate both left-sided atrial tachycardia and AF.61,63 Fibrosis and fatty infiltrates upregulate pro-inflammatory cytokines to enhance the pro-inflammatory state of the atrium, which in turn increases the likelihood of recurrent AF.64,65 These processes lead to higher AF burden and further fibrosis. AF burden has been correlated with the degree of fibrosis found in postmortem tissue: those with persistent AF had a higher degree of fibrosis than those with paroxysmal AF.66 The degree of atrial fibrosis has been associated with AF burden, AF recurrence after ablation, and cardioembolic events (as discussed later here).
The rapid atrial pacing model of AF simulates the rapid atrial rates seen in AF. In this model, animals undergo persistent rapid atrial pacing with atrial rates up to 900 BPM to induce AF. After a sustained period of time (6 weeks–180 days), the atrium remains in persistent AF once pacing is stopped.65,67 Morillo et al. first showed that sustained rapid atrial pacing led to atrial myopathy with a dilated LA.68 It is hypothesised that persistent atrial tachycardia leads to increased expression of the TGF-beta1/Smad signalling pathway with resultant atrial fibrosis as previously discussed.17 Furthermore, there are mRNA changes that lead to an activated fibroblast phenotype that leads to increased ECM.17,21 In a study of 26 sheep who underwent rapid atrial pacing with induction of AF, there was a significantly greater development of fibrotic fatty infiltrative tissue in the atria compared with those who received sham treatment.65 The development of atrial fibrosis has been noted in the rabbit and canine rapid atrial pacing models, but not uniformly.17,21,59 These findings demonstrate how persistent rapid atrial rates such as in AF may lead to atrial fibrosis. Atrial biopsies from humans with AF show similar fibrotic and fatty infiltrative changes as seen in the rapid pacing model.69–71 After multivariate analysis of clinical risk factors in subjects with atrial fibrosis, only history of AF was a significant predictor of atrial fibrosis.65 Furthermore, the degree of fibrosis was associated with AF burden.65 Rapid pacing has also been shown to induce electrical remodelling with increases in dominant frequency and ion channel density.72 These changes persisted despite mitigation of fibrosis and atrial dilation by eplerenone.72
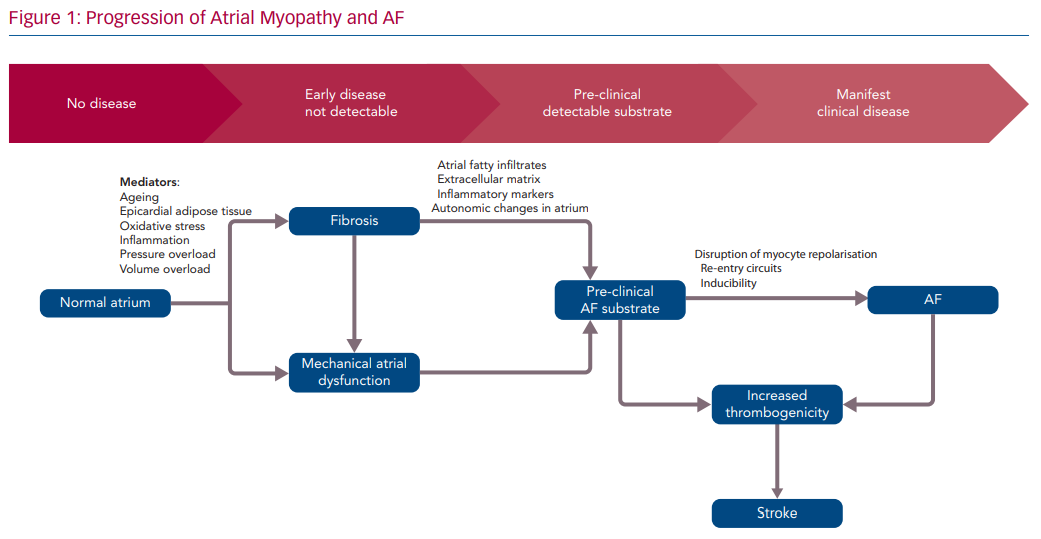
It has long been accepted that "AF begets AF".73 Due to the cyclic nature of atrial myopathy and AF, it is difficult to ascertain which develops first. However, there is a clear association between the two with a resultant positive feedback loop with downstream effects (Figure 1).74
Clinical Evaluation of Myopathy
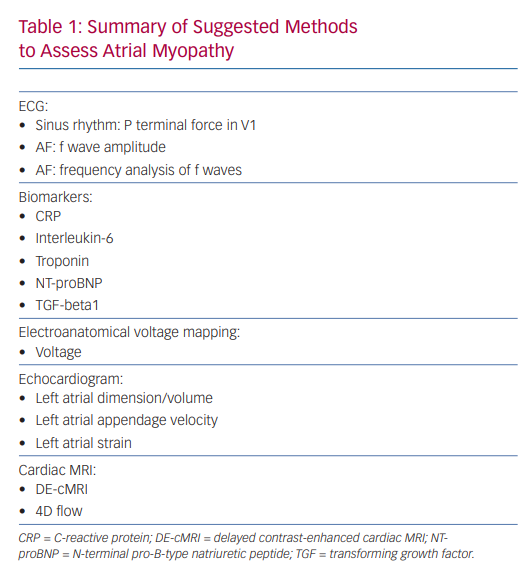
A variety of techniques have been studied to help define the atrial myopathy, including ECG, biomarkers, intracardiac voltage mapping, echocardiographic evaluation of the LA, and cardiac MRI. Here, we will explore how they can be utilised clinically to identify myopathy and potentially predict morbidity from AF and recurrence after intervention.
ECG
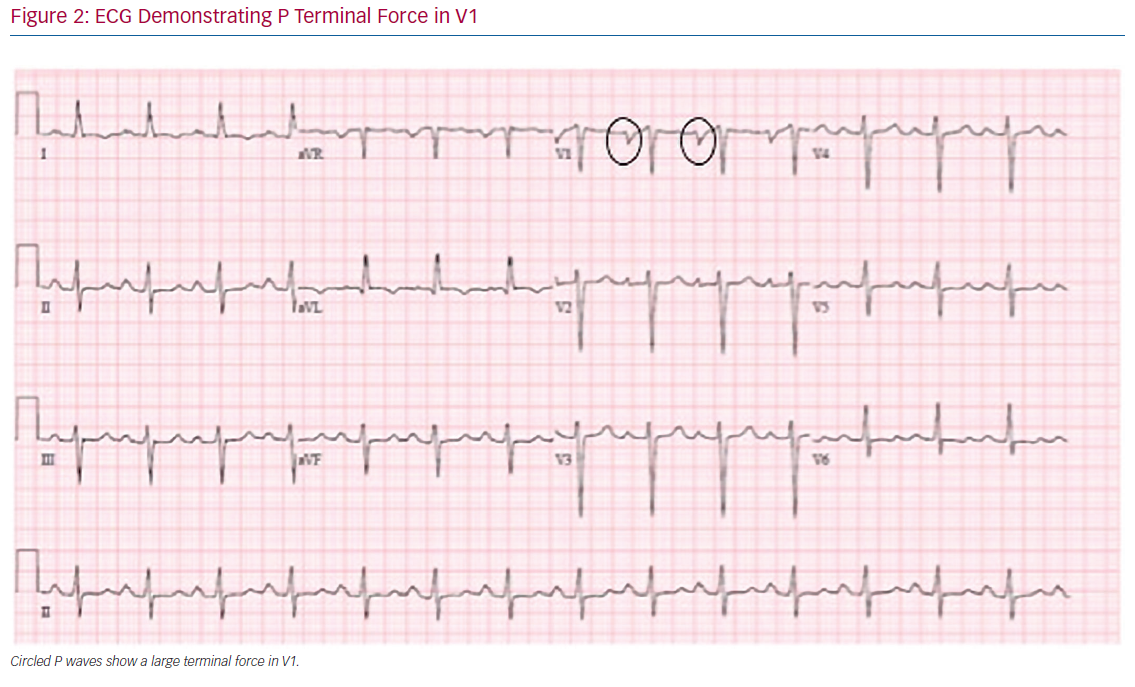
Analysis of the surface ECG during sinus rhythm is a readily available tool to predict the development of atrial myopathy and AF. The terminal force of the P wave during sinus rhythm in lead V1 (PTFV1) has been shown to correlate with LA abnormalities, such as enlargement and conduction defects (Figure 2).75,76 In a large cohort study, PTFV1 >0.06 mm/s was associated with an increased risk for the development of AF (HR 1.91; 95% CI: [1.34–2.73]; p<0.001) and with mortality (HR 1.91; 95% CI [1.34–2.73]; p<0.001).77 Ishida et al. found similar predictive abilities of PTFV1.78 Furthermore, PTFV1 has been shown to be independently associated with cryptogenic, cardioembolic, and ischemic strokes.79,80 PTFV1 ≥0.04 mm/s, along with P-wave duration ≥125 ms and P-wave dispersion ≥40 ms, have been shown to be predictors of AF recurrence after ablation.81
The fibrillatory f wave itself has been shown to correlate with the degree of LA enlargement and myopathy. The f wave amplitude has been shown to correlate with LA size and chronicity of AF (i.e. large f waves in persistent AF).82 Likewise, patients with coarse AF were more likely to have spontaneous contrast in the LAA and/or thrombus on echocardiogram.83 In addition, patients with fine AF are less likely to have AF recurrence after cardioversion compared with coarse AF.84 Frequency analysis and signal processing techniques of f waves can be predictive of clinical AF outcomes, such as response to anti-arrhythmic drugs, cardioversion, and ablation.10,85,86 Furthermore, higher f wave dominant frequency has been found in patients with persistent AF compared with paroxysmal AF, although this may be due to electrophysiologic and/or structural remodelling as AF persists.87 A recent study of patients presenting to the emergency room with a first episode of AF found no difference in dominant frequency between AF types.88 Further research is needed to establish a role for fibrillatory wave analysis.
Biomarkers
Biomarkers, although not necessarily specific to atrial pathology, have been studied both for their association with AF and AF related outcomes.45 The underlying pathophysiology represented by these biomarkers may provide an insight into the atrial myopathy. Pro-inflammatory and cardiac injury biomarkers such as C-reactive protein (CRP), interleukin-6, TGF-beta1, troponin, and N-terminal pro-B-type natriuretic peptide (NT-proBNP) have been found to be associated with the development of AF.89–92
Serum TGF-beta1, which was previously discussed as part of the fibrotic pathway, has a strong positive correlation with areas of low endocardial atrial voltage consistent with fibrosis (r2=0.93; p<0.001).93 In addition, TGF-beta1 expression is specifically upregulated in patients with extensive low-voltage areas undergoing ablation.94 Serum TGF-beta1 has been shown to be an independent risk factor for AF recurrence after ablation in both paroxysmal and persistent AF.93,95 Interestingly, Kishima et al. found lower concentrations of TGF-beta1 in patients with recurrence after ablation.96 They suggested that the discrepancy may be related to the inflammatory state of the patient. The authors concluded that in the early fibrotic phase and inflammatory stage, TGF-beta1 is higher but begins to decrease once the atrium is more heavily fibrotic.96
Additionally, high levels of the inflammatory cytokine CRP have been associated with higher risk of recurrence after cardioversion.97 Decreased aldosterone concentration has been associated with longer maintenance of sinus rhythm after cardioversion.98,99 This may reflect a reduction in the RAAS system that drives the myopathy. Elevated levels of troponin and NT-proBNP can highlight the existence of myopathy. Post-hoc analysis of the Randomized Evaluation of Long-Term Anticoagulation Therapy (RE-LY) study, showed that patients with persistent elevation of troponin and NT-proBNP were at a higher risk for cardiovascular events and mortality during the follow-up period. Likewise, data from the Aristotle trial showed that troponin levels were significantly associated with higher risk of stroke and systemic embolism.100 A recent study of circulating biomarkers associated with increased myocardial interstitial fibrosis noted a correlation with both the development of AF and recurrence after AF ablation.101
Furthermore, when added to conventional risk scoring systems, biomarkers may enhance predictive value.100,102 For example, the age, biomarkers and clinical history (ABC) stroke score calculates risk based on age, troponin/NT-proBNP levels, and prior stroke. In a validation cohort, this scoring system had a higher C-statistic of 0.65 compared with 0.60 for the CHA2DS2-VASc score.102 While not an exhaustive list, these initial studies of cardiac biomarkers highlight clinical associations that may serve as additional tools for risk stratification of patients with AF.
Intracardiac Electroanatomical Voltage Mapping
In patients undergoing electrophysiology studies, in particular those undergoing ablation procedures, electroanatomical voltage mapping can help identify the substrate for tachyarrhythmias. For AF, 3D electroanatomical voltage maps can help identify low-voltage areas that may represent areas of fibrosis (Figure 3).103 Voltage mapping can be performed in patients in sinus rhythm or AF.104 Patients with AF had significantly lower voltage in both the right atrium and LA compared with control patients with left-sided accessory pathways (right atrium: 1.7 ± 0.4 mV versus 2.9 ± 0.4 mV; LA: 1.7 ± 0.7 mV versus 33. ± 0.7 mV; p<0.001).105
Studies have shown that patients with persistent AF have a higher burden of low-voltage areas compared with those with paroxysmal AF.106 Furthermore, these low-voltage areas can be directly targeted with radiofrequency (RF) ablation to perform substrate modification. This has been reported to improve ablation success rates.106–108
Patients with a higher burden of low-voltage areas found on pre-ablation mapping are more likely to have recurrence after ablation.109–112 Vlachos et al. showed that patients with paroxysmal AF who have voltage less than 0.4 mV over more than 10% of the LA area were more likely to have recurrence after pulmonary vein isolation.113 Similar results were seen for both paroxysmal and persistent AF after ablation when using a cut-off of 0.5 mV (HR 5.89; 95% CI [2.16–16.0]; p=0.001).114–116
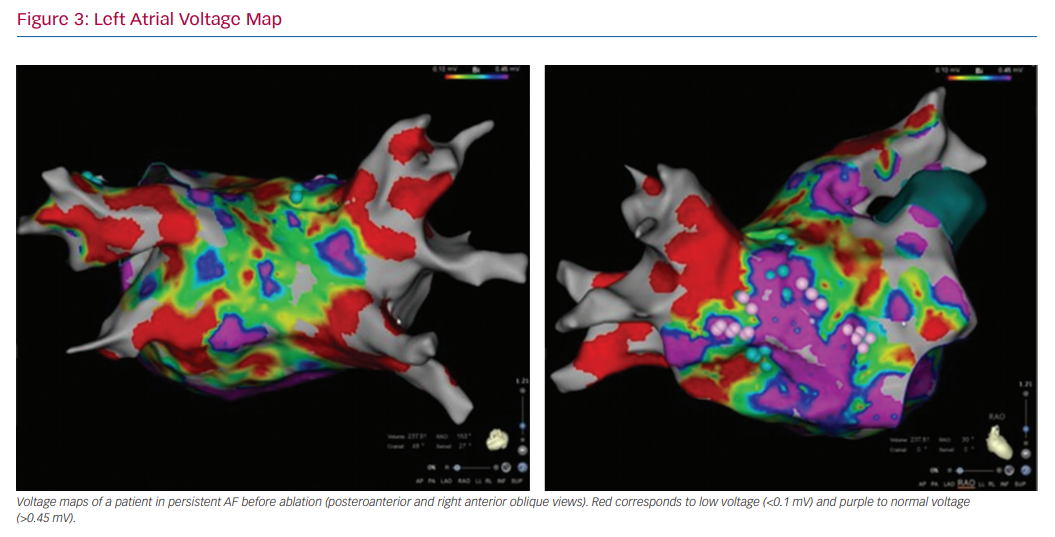
Low-voltage area burden has been associated with traditional thromboembolic risk factors such as age, female gender and LA surface area and volume.110,117 The mean LA voltage is significantly lower in patients with higher CHA2DS2-VASc scores than in those with lower scores.118 Furthermore, patients with a larger area of low voltage were more likely to have had previous stroke.119
Echocardiogram
Echocardiography allows for easy quantification of LA parameters such as diameter, area, volume, and volume index. These measurements can be used as an initial tool but they offer only a limited ability to predict the development of AF and recurrence after treatment.120–122 Furthermore, LA diameter on M-mode echocardiography did not predict stroke (RR 1.02/mm; p=0.10).123 Post-hoc analysis of the Atrial Fibrillation Follow-Up Investigation of Rhythm Management (AFFIRM) trial showed no association between LA size and the risk of stroke.124 AF patients have also been found to have LA dysfunction despite normal size.125 Therefore, current guidelines do not include LA size in the decision to start anticoagulation.7
Other echocardiographic markers are now being investigated. Diastolic early transmitral flow velocity/mitral annular velocity (E/E’; a non-invasive surrogate marker for LA end-diastolic pressure) has been found to correlate with low-voltage areas on mapping and recurrence after ablation.126 Additional information that can be obtained from transoesophageal echocardiography include LAA velocity measured on Doppler and spontaneous echocardiographic contrast, which can be predictive of future LA thrombus formation.127,128
Speckle tracking echocardiography (STE) has been proposed as a non-invasive tool to better predict atrial fibrosis.129 STE is a non-Doppler echocardiographic method to quantify atrial deformation by calculating both the atrial strain and longitudinal strain rate of atrial segments.130 Reduced atrial strain, as calculated using STE, has been correlated with reduced atrial compliance and increased fibrosis.130 Furthermore, histological analysis of patients undergoing mitral valve surgery for severe mitral valve regurgitation found a high inverse correlation between peak longitudinal atrial strain and LA fibrosis (r= −0.82, p<0.001).131
LA strain correlates with the risk of both the development of new onset AF and recurrence after treatment. In a study of patients after ST-elevation MI, multivariate analysis showed global longitudinal strain to be an independent predictor of the development of AF (HR 1.12; 95% CI [1–1.25]; p=0.042, per 1% decrease).132 Furthermore, when stratified into tertiles of longitudinal strain, patients in the lowest tertile had twice the risk as compared with the highest tertile (HR 2.10; 95% CI [1.04–4.25]).132 Patients with lower LA total strain and global strain were found to have a higher recurrence of AF after ablation (OR 0.81; 95% CI [0.73–0.89]; p<0.0001).133
LA strain on STE correlates with the CHADS2 score.134 Moreover, there was an improvement in the prediction of hospitalisation and mortality when LA strain and indexed LA volume were combined with the CHADS2 score (p=0.003).134 In patients with permanent AF, peak strain had an independent negative correlation with prior stroke when controlling for age, LA volume index, and ejection fraction.135 Overall, these examples of STE highlight the interplay between atrial fibrosis and AF given that lower strain has been consistently correlated with AF severity and morbidity.
Cardiac MRI
Delayed contrast-enhanced cardiac MRI (DE-cMRI) has been applied to the assessment of atrial fibrosis.136 Although its use in the left ventricle for scar detection is widespread, it is much more challenging to apply to the atrium due to its thinner wall and the limited resolution of MRI.130,137 Cardiac MRI is performed by analysing the distribution of gadolinium contrast agent 10–15 minutes after it is administered. In normal cardiac tissue, the contrast agent quickly washes out. However, in fibrotic tissue it accumulates in the ECM.136,138 A positive correlation has been shown between areas of delayed enhancement and areas of low voltage in pre-ablation atrial mapping (r2=0.61).139 A similar association between cardiac MRI and voltage mapping was seen by Spragg et al.137 The Utah classification scheme has been proposed to quantify LA fibrosis using DE-cMRI. Based on this system, patients are assigned to 4 groups: Utah I, defined as ≤5% LA wall enhancement; Utah II, 5–20%; Utah III, 20–35%; and Utah IV, >35%. This schema has been shown to be useful in predicting recurrence of AF after catheter ablation in both lone AF and non-lone AF.140,141
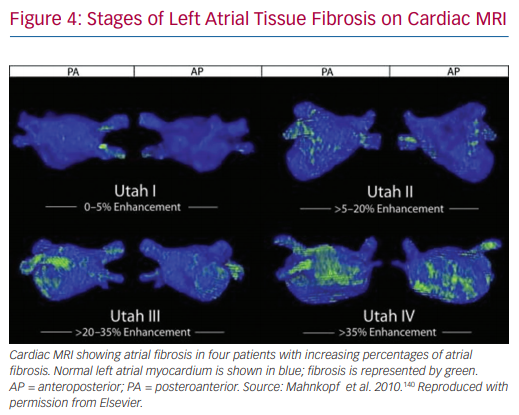
The DE-MRI Determinant of Successful Radiofrequency Catheter Ablation of Atrial Fibrillation (DECAAF) study is a large multicentre prospective trial that analysed the ability of the Utah criteria to predict recurrence after ablation.141 This study showed an independent association between MRI-detected atrial fibrosis and recurrent arrhythmia. The hazard ratio was 1.06 (95% CI [1.03–1.08]) for every 1% increase in LA fibrosis. Furthermore, cumulative incidence of recurrence after 325 days based on Utah score was 15.3% (95% CI [7.6–29.6%]) in Utah I, 32.6% (95% CI [24.3–42.9%]) in Utah II, 45.9% (95% CI [35.5–57.5%]) in Utah III, and 51.1% (95% CI [32.8–72.2%]) in Utah IV (Figure 4).141
Similar results have been found when using DE-cMRI to evaluate fibrosis to predict thromboembolic risk. A recent retrospective study of more than 1200 patients who had undergone DE-cMRI evaluated the association of Utah stage atrial fibrosis with cerebrovascular events. It found a significant association between higher late gadolinium enhancement and the rate of cerebrovascular events (HR 3.94; 95% CI [1.72–8.98]).142 An additional analysis of the Utah dataset found that women had a higher rate of fibrosis than men (17.5% versus 15.3%; p<0.01) which may explain the increased stroke risk in women with AF (prior stroke prevalence 15.8% versus 6.5%; p<0.001).143 Moreover, in a prospective study of 111 patients with ischemic stroke, patients with a cryptogenic stroke after extensive stroke workup were found to have a statistically significantly higher percentage of LA fibrosis than patients with a known cause excluding AF (LA fibrosis percentage: median 18%, IQR 16% versus median 10.5%, IQR 16%; p=0.03). Patients with an unknown cause had a similar percentage of fibrosis compared with those with AF as cause (median 18%, IQR 16% versus median 25%, IQR 21%; p=0.22).144
Additionally, 4D flow cardiac MRI, in which 3D images are processed over time, allows for the quantification of LA velocities.145 Subjects with AF had lower velocities than age-matched controls, and those imaged in AF had lower velocities than AF patients imaged in sinus rhythm.145 While not a direct assessment of fibrosis, 4D flow MRI provides a surrogate index for the extent of the atrial myopathy. Reduced LA velocities have been associated with increased stasis and increased CHA2DS2-VASc score (p<0.043).146–148
Future directions for cMRI include 3D segmentation, quantification, and visualisation of LA fibrosis and T1 mapping of the LA to measure extracellular volume as a surrogate marker for LA fibrosis.149,150 However, further research needs to be done to validate these techniques and their clinical utility (Table 1).
Guiding Therapies to Target Fibrosis
Current therapies such as anti-arrhythmic drugs and catheter ablation do not target the atrial myopathy that serves as the substrate for AF. Although catheter ablation of the pulmonary veins has a Class I indication for symptomatic AF,7 extensive RF ablation generates additional scar in the atrium. While this scar tissue may remove arrhythmogenic areas, new extensive scarring may have untoward effects. Patients after RF ablation often have decreased LA area and volume, which may be the result of scar formation with resultant decreased compliance.151,152 Alternatively, this may be due to reverse remodelling of the atrial myopathy. Using cardiac MRI, a correlation was seen between both the number of ablation procedures (r=0.58, p=0.002) and RF duration with the degree of post-ablation atrial scar (r=0.56, p=0.003). Additionally, post-ablation LA scar was negatively correlated with LA compliance and active LA ejection fraction.153 Similar findings were seen in a study by Wylie et al.154 Those authors concluded that scar will lead to a further decrease in LA compliance, which can lead to “stiff LA” syndrome.154
Conversely, prevention of progression and even regression of atrial myopathy has been reported after catheter ablation. There has been some evidence of atrial remodelling following catheter ablation in patients with baseline cardiomyopathies. In a subset of patients from the Catheter Ablation Versus Medical Rate Control in Atrial Fibrillation and Systolic Dysfunction (CAMERA-MRI) study, repeat electroanatomical mapping after pulmonary vein and posterior wall isolation by ablation showed an increase in right atrial voltage on repeat electroanatomical mapping at a mean time of 23.4 months after index ablation (from 1.6 mV ± 0.1 mV to 1.9 ± 0.1 mV; p=0.04). This along with reduction in atrial size and complex fractionated electrograms (21.7 ± 3.5% to 8.3 ± 1.8%; p=0.002) may indicate possible reversal of the atrial myopathy.155 This reverse remodelling may be the result of maintenance of sinus rhythm after ablation.
There are preliminary results suggesting that therapies such as weight loss, angiotensin receptor blockers, aldosterone inhibitors, and statins may be able to reduce fibrosis to prevent and decrease AF burden.156–161 For example, patients treated with angiotensin II receptor antagonists were found to have lower levels of TGF-beta1 than those not treated.162 Clinically, the Routine Versus Aggressive Upstream Rhythm Control for Prevention of Early Atrial Fibrillation in Heart Failure (RACE III) trial randomised patients with persistent AF and mild–moderate heart failure to aldosterone antagonist, ACE inhibitors, statins, and cardiac rehab versus conventional therapy. At 1 year, sinus rhythm was present in 75% of the intervention group compared with 63% in the control group (OR 1.765; p=0.042).163 There was also a decrease in markers of myopathy such as NT-proBNP. Similarly, in the Eplerenone in Mild Patients Hospitalization And SurvIval Study in Heart Failure (EMPHASIS-HF) trial, patients with systolic heart failure randomised to eplerenone had decreased incidence of new-onset AF (2.7% versus 4.5%; HR 0.58; 95% CI [0.35–0.96]; p=0.034).164 Eplerenone has been shown in the sheep model to reduce atrial fibrosis and dilation, AF inducibility, and progression to persistent AF without preventing AF-induced electrical remodelling.72 In the Aggressive Risk Factor Reduction Study for Atrial Fibrillation and Implications for the Outcome of Ablation (ARREST-AF), patients were randomised to risk factor modification guidelines as recommended by the American Heart Association/American College of Cardiology (AHA/ACC) versus conventional care. The treatment arm had an increase in arrhythmia-free survival rates as compared with the control (32.9% versus 9.7%; p<0.001) with HR of 2.3 (p<0.001). While the mechanism is unclear, improvements in atrial volume on echocardiography suggest that it may be due to reversal of the underlying cardiomyopathy.165 As a result, the latest ACC/AHA guidelines for AF recommend weight loss in obese patients with a Class I indication.7 Additionally, Kunamalla et al. demonstrated the use of targeted gene-based reduction of TGF-beta signalling to decrease fibrosis and AF in the canine model.166 Other potential targets of gene therapy include modification of ion channels, gap junctions, autonomic innervation, and fibrosis.167
Conclusion
This review highlights the complex interplay between atrial myopathy and AF incidence, recurrence, burden, and stroke risk. Atrial myopathy consists of multiple changes to the atrium including increased fibrosis, pressure overload, autonomic derangement, endothelial dysfunction, and alterations in prothrombotic factors. While there are currently multiple modalities to assess atrial myopathy, they are not being utilised routinely when making decisions regarding therapy including anticoagulation. The additional use of these modalities may allow us to better predict outcomes and tailor therapy. Creating novel therapies that address inflammation, EAT, the autonomic nervous system and molecular pathways of atrial fibrosis are an exciting new paradigm for future AF treatment. Reducing and even reversing atrial myopathy may serve as a more durable method to treat AF and reduce its significant morbidity. Further research is needed to develop these novel strategies to address atrial fibrosis as the precursor to atrial myopathy, AF, and cardioembolic stroke.
Clinical Perspective
- AF and atrial myopathy form a complex cycle in which each contributes to the other.
- Current risk stratification schema and management strategies do not consider the underlying myopathy of AF.
- By identifying atrial myopathy, we may be able to better risk stratify and treat patients with AF.