










The Risk of Atrial Arrhythmias in Inherited Primary Arrhythmia Syndromes
The inherited primary arrhythmia syndromes (IPAS) are a heterogeneous group of diseases caused by mutations in genes encoding for cardiac ion channels. People affected by one of these inherited diseases have no overt structural cardiac abnormalities but are at higher risk of sudden cardiac death due to the occurrence of life-threatening ventricular arrhythmias.1 Cardiac channelopathies include Brugada syndrome (BrS), short and long QT syndromes (SQTS and LQTS), early repolarisation syndrome (ERS) and catecholaminergic polymorphic ventricular tachycardia (CPVT).1 The vast majority of IPAS present with a specific ECG phenotype, characterised by an abnormal ventricular depolarisation and/or repolarisation phase, such as the coved-type ST-segment elevation in the right precordial leads, infero-lateral early repolarisation (ER), long QT and short QT intervals.1,2 These abnormalities reflect altered imbalance in ionic currents of cardiac myocytes, that may lead to development of ventricular arrhythmias such as monomorphic, polymorphic ventricular tachycardia (VT) or VF.
Since the diagnostic ECG ventricular pattern of many IPAS is dynamic and often concealed, it is particularly difficult to estimate the prevalence of these diseases in the general population. However, most studies indicate that BrS is probably the most common with a prevalence ranging from 1:2,000 in Europe to 1:1,000 in Asian and south-east Pacific countries, followed by LQTS which has an estimated prevalence of four in 10,000 worldwide.1 BrS may present overlapping ECG and clinical features with ERS and an ER pattern in the infero-lateral leads with a J-point elevation ≥0.2 mV has been observed in up to 6% of community-based middle-aged subjects without BrS.3 SQTS and CPVT are the two rarest cardiac channelopathies with a prevalence of 2.7 in 100,000 and 1 in 10,000, respectively, worldwide.1
Both atrial and ventricular cardiomyocytes may express proteins encoded by the same mutated gene, which makes atrial arrhythmias common in patients with inherited primary arrhythmia syndromes.4 Paroxysmal AF is by far the most common atrial arrhythmia observed in these patients, and is occasionally the first and only manifestation of an inherited cardiac ion channel dysfunction.2,5
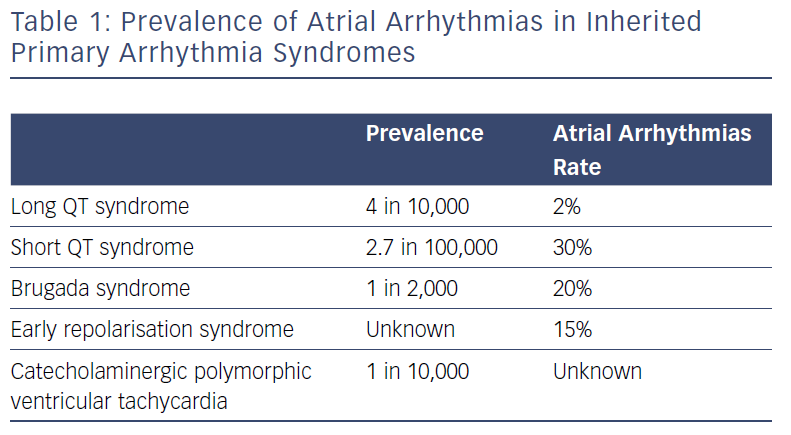
While the prevalence of paroxysmal AF in the young (aged <50 years) is 0.1%, prevalence of AF is significantly higher among people with IPAS, ranging from 2% for patients with genetically proven LQTS to 30% for subjects with SQTS (Table 1).6–8
In recent years, scientific evidence has been provided on the genetic basis of AF in the general population. Many monogenic mutations or rare variants have been identified.14 However, although useful in understanding the pathophysiology of AF, these mutations or rare variants have limited value in explaining the heritability of AF, accounting only for sporadic or familial cases of AF. Genome-wide association studies for AF have identified 97 loci that are significantly associated with an increased risk of AF that might be potential signals for the causative genes.14 It is still unknown the specific role of genetic variants in enhancing the risk of atrial arrhythmias in IPAS.
Long QT Syndrome
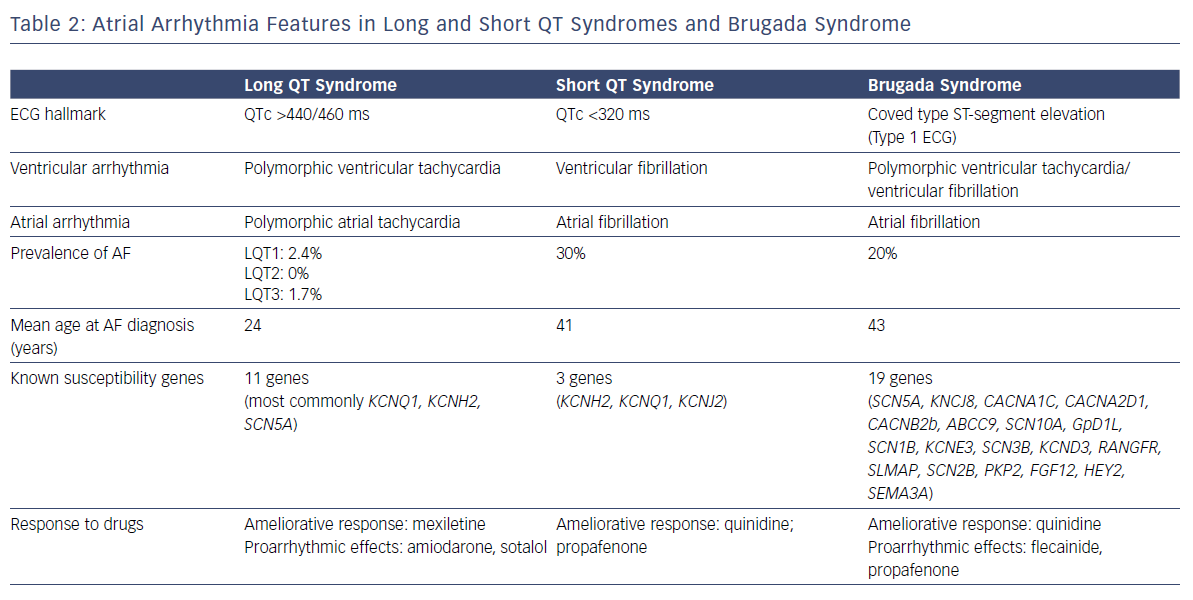
Eleven LQTS susceptibility genes have been described so far. However, mutations in three genes (KCNQ1-mediated LQT1, KCNH2-mediated LQT2, and SCN5A-mediated LQT3) account for 75% of clinically definite LQTS. It has been shown that a patient with LQTS has a 17-fold higher risk of experiencing AF before the age of 50 compared with population-based individuals (RR 17.5, CI 2.2–139.6).7 Of patients with genetically proven LQTS and AF, 63% present with a loss-of-function mutationin KCNQ1.7
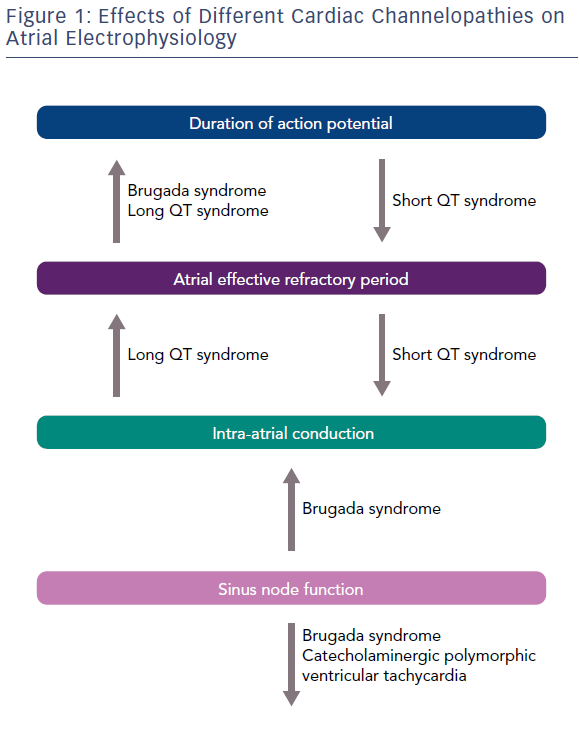
Kirchhoff et al. reported that patients with LQTS have altered atrial electrophysiology due to the prolongation of the atrial action potential and the atrial effective refractory period (Figure 1). These alterations, possibly determined by a dysfunction of KCNQ1-encoded IKs, lead to an enhanced susceptibility for an atrial form of torsades de pointes, having a longer cycle length, but resembling AF on the surface ECG (Table 2).9 Similarly, Satoh and Zipes reported the effects of caesium chloride, a potassium channel blocker, in determining polymorphic atrial tachyarrhythmias by producing early after-depolarisations in canines. Based on these observations, it might be speculated that patients with LQTS present a similar arrhytmogenic platform for the development of both ventricular and atrial tachyarrhythmias.10
Short QT Syndrome
Three gain-of-function gene mutations encoding for cardiac potassium channels have been associated with SQTS: KCNH2 (SQT1), KCNQ1 (SQT2) and KCNJ2 (SQT3). AF is common in patients with SQTS and in half of all cases it is the first clinical manifestation of the syndrome.8 The potential physio-pathological mechanism responsible in SQTS for both atrial and ventricular arrhythmias is shortening of the refractory period due to a gain of function of potassium channels.11
Early Repolarisation Syndrome and Catecholaminergic Polymorphic Ventricular Tachycardia
No specific information is available on atrial arrhythmias in patients with ERS and CPVT. Junttila et al. reported that paroxysmal AF occurs in 15% of middle-aged subjects with an infero-lateral ER pattern.12 A gain-of-function in gene encoding in the cardiac potassium channel (Kir6.1) is associated with both increased AF susceptibility and ER, indicating a role for the ion channel function in both ventricular and atrial repolarisation.13 Ventricular pathophysiological mechanisms leading to ERS are still controversial and no specific studies have been carried out to assess the atrial electrophysiological abnormalities in these patients.
Atrial tachyarrhythmias seem to be rare in patients with CPVT. Exercise-induced supraventricular arrhythmias have been only sporadically reported in patients with CPVT and no further specific data have been reported.1,4 More than half of CPVT cases are caused by autosomal-dominant missense mutations in the ryanodine receptor (RyR2), which is the major calcium (Ca2+) release channel on the sarcoplasmic reticulum (SR) required for excitation-contraction coupling in cardiac muscle.15 In mouse models of CPVT, it has been shown that RyR2-mediated diastolic SR Ca2+ leak in atrial myocytes is associated with AF.16 Moreover, most experimental studies agree that RyR2 phosphorylation by protein kinase A and Ca2+ calmodulin-dependent protein kinase II can potentiate SR Ca2+ leak, which is associated with ectopic activity and susceptibility to arrhythmias in CPVT.17
Atrial Arrhythmias in Brugada Syndrome
The ECG hallmark of BrS is the presence of a coved-type ST-segment elevation of >2 mm (Brugada type 1 ECG) in at least one right precordial lead (V1 and V2), placed in a standard or superior position.2 The Brugada type 1 ECG can be concealed, becoming apparent only under certain conditions such as fever or enhanced vagal tone. Most prominent ECG changes usually appear just before the onset of ventricular arrhythmias. The Brugada type 1 ECG is required for the diagnosis of BrS and, in the case of a non-diagnostic baseline ECG, can be unmasked by the IV administration of sodium channel blockers, such as ajmaline or flecainide.2
The ECG phenotypic expression of BrS is determined by the presence of an outward shift of the balance in repolarising currents at the level of the right ventricular outflow tract (RVOT), caused by a decrease in sodium or calcium channel currents or an increase in outward potassium currents, which creates a notch in the action potential of the epicardium resulting in a transmural voltage gradient.18 Although the pathophysiological origin of the syndrome is still under debate, there is general consensus that the presence of functional abnormalities in the epicardial RVOT leads to the ventricular phenotypic manifestation of the disease. Nademanee et al. observed that abnormal fractionated late potentials can be found in the RVOT epicardium of people with BrS and can be eliminated by catheter ablation.19 These findings were later confirmed by Pappone et al., who reported that epicardial mapping and ablation of BrS can be safely performed and its efficacy can be confirmed by post-ablation remapping and flecainide testing, and results in the elimination of abnormal substrate and simultaneous disappearance of the typical BrS ECG pattern and no further inducibility of VT/VF.20
Pharmacological therapy with quinidine together with ICD implantation can be used as an alternative to catheter ablation in high-risk patients.21 The efficacy of quinidine is based on its blocking effects on Ito currents (transient outward potassium currents) and can be tested by the absence of VT/VF inducibility during programmed ventricular stimulation.21 Moreover, it has been recently reported in a series of 20 BrS patients undergoing RVOT endomyocardial biopsy that pathologic findings with myocardial inflammation were present in 75% of cases, which prompts the question of whether a combination of electrical and structural abnormalities can explain the arrhythmogenic substrate of BrS.22
Among people with IPAS, BrS is the most genetically heterogeneous disease and has a considerable allelic heterogeneity: mutations in different genes can, in fact, lead to the same clinical manifestation and different mutations in each gene can eventually cause the disease. In addition, different gene mutations and variants can coexist and affect one or more subunits of the potassium, calcium and sodium channel structures.23 BrS has been associated with mutations in 19 different genes. Genetic abnormalities are found in up to 30% of genotyped patients with BrS.24
Paroxysmal AF is the most common supraventricular arrhythmia in patients with BrS, occurring in about 20% of cases.2,25 Paroxysmal AF represents a relevant clinical issue in BrS for several reasons. Patients with BrS who develop paroxysmal AF have a more severe phenotype and advanced disease process with higher incidence of syncopal episodes, spontaneous or induced sustained ventricular arrhythmias and spontaneous Brugada type 1 ECG.26,27 The incidence of AF is higher in patients with an ICD and up to 20% of these patients can experience inappropriate shocks because of AF with rapid ventricular response.28 Appropriate screening and the choice of the most appropriate type of device (single, dual-chamber or subcutaneous), together with careful programming, is necessary to correctly detect episodes of fast AF and reduce the rate of inappropriate shocks, which are significantly affecting the quality of life of these generally young patients. Moreover, patients with BrS who develop AF have a more frequent positive genetic test, but SCN5A mutation has been associated with prolonged atrial conduction time and AF induction at EPS, but not disease severity.29
The pharmacological treatment of paroxysmal AF in BrS is limited by being unable to use conventional AADs with sodium channel blocking properties, which might be pro-arrhythmic, exposing patients to the development of life-threatening arrhythmias. Indeed, according to the brugadadrugs.org website, flecainide, ajmaline, procainamide, pilsicainide, propafenone are drugs that should be strongly avoided because they have been associated with an enhanced risk of ventricular arrhythmias. Amiodarone, propanolol, verapamil and vernakalant should be preferentially avoided, while there is no mention on the use of sotalol, quinidine or other beta-blockers. However, the latter are underused and not well tolerated by these young patients due to their side-effects.
Although pulmonary vein (PV) isolation is currently the most widespread ablation strategy to treat patients with paroxysmal AF, the choice of the best invasive treatment in patients with BrS remains controversial. Indeed, in the general population, pathophysiological mechanisms leading to AF include adverse remodelling of the atrial electro-anatomical substrate and/or abnormal triggered activity.30 Pulmonary vein ectopic activity has been established to be the main trigger of paroxysmal AF in patients without structural heart disease.31 Conversely, the pathophysiological mechanisms of AF in patients with BrS are still unknown and the role of PV triggering remains uncertain.32 Moreover, long-term results of AF ablation in patients with BrS are relatively poor when compared with similarly aged with lone AF.33 In fact, at a mean follow-up time of 22 months, nearly 65% of BrS patients were free from AF recurrences after having PV isolation.
The arrhythmogenic substrate of BrS might not be restricted to the ventricular level. Similar atrial changes could be the substrate for reentrant atrial tachyarrhythmias but have not been extensively studied. The presence of a prominent transient outward current in atria and the observation that episodes of AF are triggered by closely coupled atrial extrasystoles points to the possibility that a substrate similar to that responsible for ventricular arrhythmogenesis may also be responsible for the development of AF in patients with BrS.34 As for ventricular arrhythmias, the onset of AF in these patients is often preceded by fluctuations in autonomic tone and most AF episodes occur at night.29 Vagal stimulation might reduce atrial conduction velocities and shorten the effective refractory period facilitating the induction of AF.29,30
Apart from AF, BrS can coexist with other forms of atrial arrhythmias, including atrioventricular nodal reentrant tachycardia (AVNRT). Indeed, a concealed BrS ECG is present in 27% of patients presenting with AVNRT.35 In these cases, genetic variants known or suspected to cause loss of function of sodium channel current may provide a mechanistic link between AVNRT and BrS and predispose to the expression of both phenotypes.35
Assessment of Atrial Electrical Activity in Patients with Brugada Syndrome
The baseline ECG of people who are prone to AF is easily obtainable and non-invasive and might reflect subclinical structural atrial abnormalities resulting from adverse remodelling of the atrial electro-anatomical substrate.36,37 The P-wave on surface ECG represents atrial depolarisation and its duration may be a reliable non-invasive measurement of atrial conduction time. Previous studies have indicated that abnormal P-wave axis and long P-wave duration are strongly associated with an increased risk of AF.38,39 Moreover, P-wave indices which include a summary measure of duration, area and amplitude have been proposed to quantify the atrial electrical properties derived from the ECG, but they have shown only limited value in predicting AF occurrence.40
The baseline ECG of patients with BrS reflects atrial electrical abnormalities resulting from the presence of an atrial channelopathy independently from the presence of previous episodes of AF. Patients with BrS have a longer P-wave duration compared with healthy controls.41,42 This anomaly might be due to the prolonged atrial action potential and increased intra-atrial conduction time reported by Kusano et al.25 These researchers did not find any significant difference in electrophysiological parameters, such as intra-atrial conduction time, duration of local atrial electrograms and atrial effective refractory period between BrS patients with type 1 and those with type 2 or type 3 determined by ECG. This shows the potential existence of a miscorrelation between the atrial and ventricular ECG phenotype, due to a different location or density of the imbalanced ion channels between atria and RVOT.
Recently, the existence of a concealed abnormal atrial phenotype has been documented in BrS without history of AF.43 Indeed, BrS patients without history of AF present with marked atrial electrical abnormalities which are even more pronounced when compared with those displayed by patients with paroxysmal AF. Moreover, the abnormal P-wave parameters can be detected even in the absence of an overt ventricular ECG phenotype, symptoms or SCN5A gene mutation, indicating the existence of a constant abnormal atrial phenotype and the presence of an atrial-ventricular phenotypic mismatch. The normal action potential in atria differs from that of the ventricle with respect to ion channel currents that contribute to resting membrane potential, phase 1 and phase 3 of the action potential.44
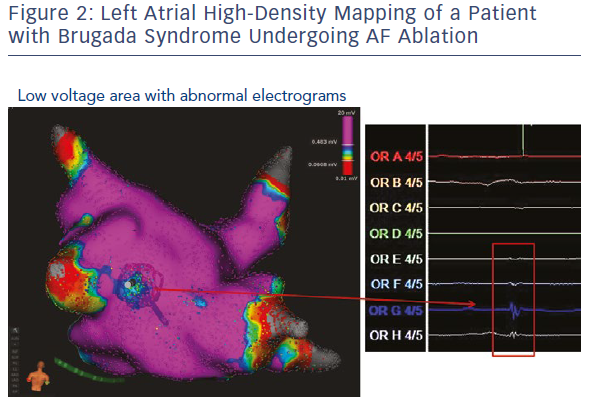
Experimental and clinical evidence suggests that there is heterogeneity of sodium currents among atrial and epicardial ventricular myocytes.44,45 Indeed, sodium current density is much greater in atrial versus ventricular cells and time constants for activation and inactivation are more rapid in atrial myocytes. These structural and pharmacological differences between atrial and ventricular sodium channels might be exaggerated in patients with an inherited sodium channel dysfunction and might explain the presence of a constantly abnormal atrial phenotype in people with BrS. High-density atrial endocardial mapping might be a useful tool to assess the presence of atrial electrical abnormalities in patients with BrS and documented atrial arrhythmias (Figure 2).
Conclusion and Future Perspectives
Available scientific data highlights the evidence that the arrhythmogenic substrate of an inherited ion channel dysfunction is not restricted to the ventricular level and similar abnormalities affect the atrial electrophysiological properties creating the substrate for the occurrence of reentrant atrial tachyarrhythmias. However, specific data on the involvement of the pathogenic process at the level of the atria are lacking and the relationship between the atrial and ventricular phenotype has not been deeply investigated in IPAS.
A modelling approach to understanding inherited primary electrical disorders at the atrial level and detailed information about three-dimensional distribution of atrial action potentials in people with cardiac channelopathies is of paramount importance. A full insight into atrial involvement in inherited primary arrhythmia syndromes provided by high-resolution ECG and mapping systems, and cardiac simulations might enrich our understanding of the pathophysiological mechanisms underlying the development of atrial arrhythmias in these patients. In the last decade, particular attention has been focused on the development of atrial-selective drugs for the prevention of AF with the goal of avoiding ventricular pro-arrhythmic effects.45,46 While atrial-selective sodium channel blockers with preferential atrial activity, such as ranolazine, might be useful in the management of AF in the general population, different effects of these drugs might be revealed in patients with a cardiac channelopathy. Indeed, the use of atrial selective sodium channel blockers may be pro-arrhythmic in BrS patients and may, eventually, lead to the development of AF.
In-depth characterisation of pathogenesis of atrial arrhythmias in IPAS might have significant clinical implications in the diagnostic and therapeutic management of a much larger group of individuals at risk of AF and, in general, in a very large group of patients affected by lone atrial fibrillation or other cardiac causes.
Clinical Perspective
- The arrhythmogenic substrate of an inherited ion channel dysfunction is not restricted to the ventricular level and similar abnormalities affect the atrial electrophysiological properties creating the substrate for the occurrence of atrial tachyarrhythmias.
- Patients with Brugada syndrome present with a concealed abnormal atrial phenotype that can be detected even in the absence of an overt ventricular phenotype.
- In-depth characterisation of pathogenesis of atrial arrhythmias in patients with cardiac channelopathies have significant clinical implications in the diagnostic and therapeutic management and lead to a better understanding of the pathophysiological mechanisms and to a more appropriate individualised treatment.