







Most mutations of the LMNA gene affect the heart, causing a dilated cardiomyopathy, ususally with conduction defect and ventricular arrhythmia, with or without skeletal muscle involvement. Although a relatively rare disease, cardiologists should be aware of laminopathies (diseases caused by LMNA gene mutations) because of the particularly aggressive course compared with most other cardiomyopathies, and because of the benefit of early defibrillator and pace maker implantation.
What are Lamin Proteins?
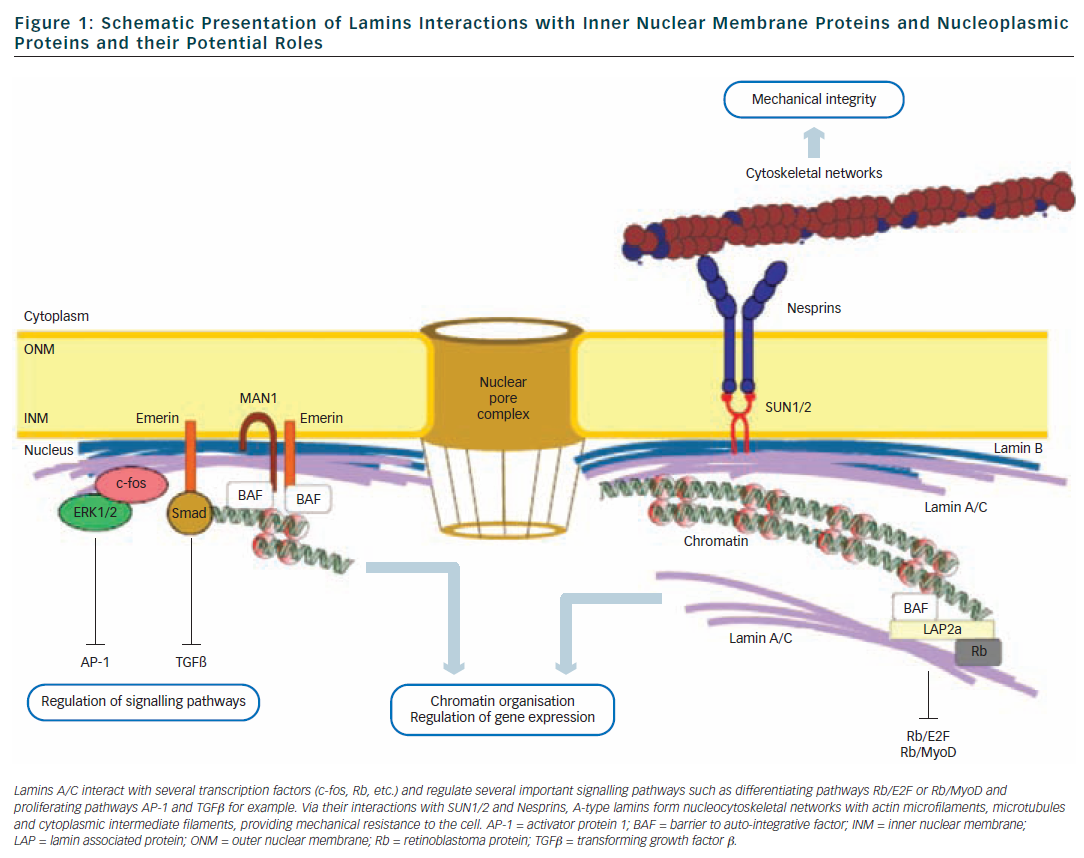
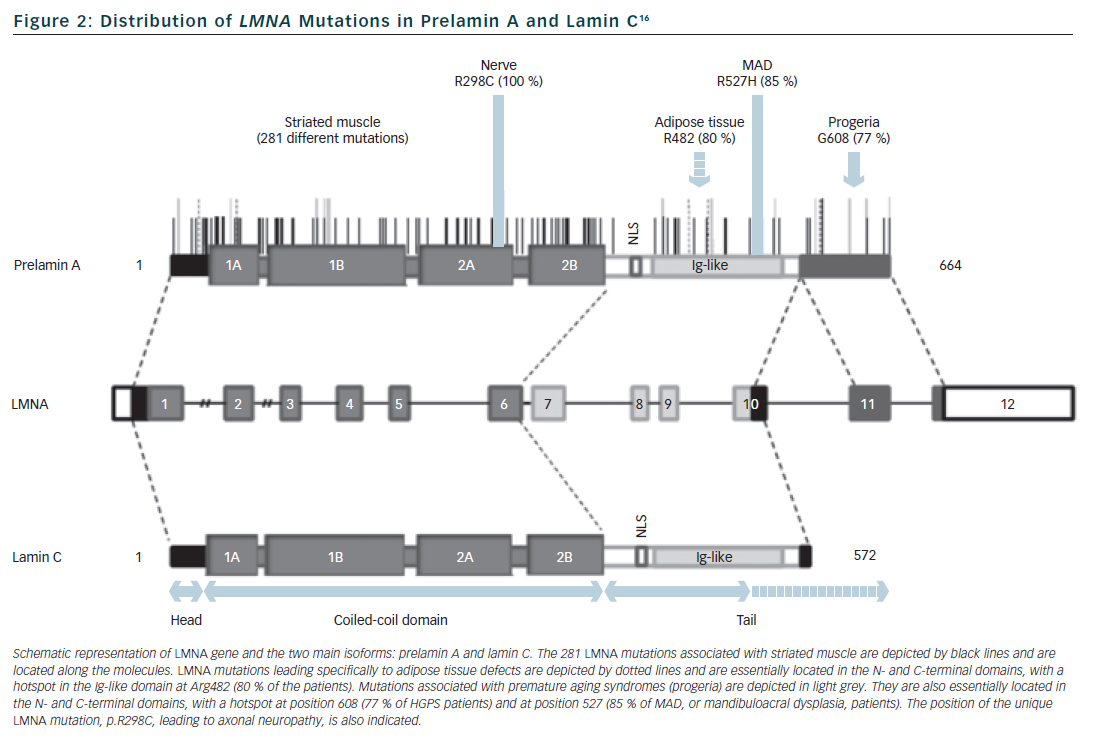
Lamins are type V intermediate filament proteins that are able to polymerise and form the nuclear lamina, an organised meshwork that lies between the inner nuclear membrane and the chromatin (see Figure 1).1–6 A-type lamins, i.e. the lamin A and the lamin C, together with B-type lamins, are the constituents of the nuclear lamina and lamin A and C isoforms are both encoded by the LMNA gene via alternative splicing (see Figure 2). The LMNA gene is localised to chromosome 1q21.2-q21.3, spans approximatively 24 kb and is composed of 12 exons that encode four lamin isoforms (A, AΔ10, C and C2). The two major isoforms lamin A and C are identical for their first 566 amino acids but differ by their C-terminal domains.7 Lamin A is initially synthesised as a precursor, prelamin A, with 98 unique C-terminal amino acids. Prelamin A is farnesylated on the cysteine residue of a C-terminal CaaX box and then is endoproteolytically processed by the ZMPSTE24 protease (zinc metalloprotease Ste24 homologue) removing the last 18 amino acids to yield the mature lamin A (74 kDa). Lamin C (65 kDa) has six unique C-terminal amino acids and is not post-translationnaly modified by farnesylation. Lamin A and lamin C (henceforth referred to as lamin A/C) are expressed in terminally differentiated somatic cells but are lacking from early embryos. In contrast, B-type lamins that are encoded by different genes (LMNB1 and LMNB2) are also present in undifferentiated cells.
The central rod domain of lamins is a highly conserved alpha-helical core of approximatively 360 residues that drives the interactions between two lamin protein chains to form a coiled-coil dimer. Lamins A/C further assemble to form head-to-tail polymers that, together with B-type lamins, constitute the nuclear lamina. One of the functions of the lamina is to provide structural support to the nucleus and maintaining the mechanical integrity of cells by linking the nucleoskeleton to the cytoskeleton.8 Other studies also support a complex role of lamins in nuclear pore function, chromatin organisation, DNA replication and transcriptional regulation9,10 (see Figure 1).
Lamin A/C Mutations and Overview of the Various Phenotypes
More than 450 different mutations have been identified in the LMNA gene and they can cause a wide variety of distinct and disparate diseases involving striated muscle (dilated cardiomyopathy, skeletal myopathies), adipose tissue (lipodystrophy syndromes), peripheral nerve (Charcot-Marie-Tooth neuropathy) or multiple systems with accelerated ageing (progerias).
Laminopathies of the Striated Muscle
The laminopathy story began in 1999 when we identified the first mutation of the LMNA gene,11 in patients affected with autosomal dominant Emery-Dreifuss muscular dystrophy (EDMD), characterised by a triad of early tendon contractures (elbows, Achille’s tendons, spine), muscle weakness/wasting with a predominantly humero-peroneal distribution, and dilated cardiomyopathy/conduction defect. Subsequently, LMNA mutations were identified in patients with dilated cardiomyopathy and conduction defect (DCM-CD), similar to EDMD, but with minimal or no skeletal muscle involvement.12 Shortly afterwards, LMNA mutations were reported in patients with limb-girdle muscular dystrophy type 1B (LGMD1B),13 which share the cardiac features of EDMD but muscle weakness/wasting predominantly involving pelvic and scapular girdle muscles, and no or mild tendon contractures. More recently, LMNA mutations were identified in congenital forms of muscular dystrophy (L-CMD) with onset before two years of age and evoluation towards severe respiratory insufficiency.14
Laminopathies of Other Tissues
The main entity of adipose tissue laminopathies is familial partial lipodystrophy of Dunnigan type (FPLD) characterised by abnormal distribution of subcutaneous fat (loss at extremities and accumulation in neck and face), metabolic syndrome with insulin-resistance, hypertriglyceridaemia and sometimes type 2 diabetes. Peripheral nerve laminopathy is related to a recessive form of Charcot-Marie-Tooth axonal neuropathy, characterised by distal muscle wasting/weakness and absence of osteotendinous reflexes due to axonal degeneration. Accelerated ageing laminopathies are mainly represented by Hutchinson-Gilford progeria syndrome, an extremely rare disorder with segmental premature ageing sparing the brain, with subjects dying at a mean age of 13 years from cardiovascular disease (atherosclerosis). Other rare or overlapping syndromes have been linked to LMNA mutations including mandibuloacral dysplasia (MAD), atypical Werner syndrome and restrictive dermopathy.15
Laminopathies and Genetic Heterogeneity
Apart from the large phenotypic pleiotropy there is also a large genetic variability with more than 450 different mutations identified in the LMNA gene, all published ones being available within the UMD-LMNA mutation database at www.umd.be/LMNA. All types of mutations are reported:16 missense mutation that is the most frequent mechanism (72 % of the 301 first published LMNA mutations), in-frame insertion/deletion (9 %), out-of-frame insertion/deletion (9 %), splice-site (7 %) and nonsense (5 %) mutations. Genotype-phenotype relationships are incompletely understood. However mutations that affect tissues other than striated muscle are usually related to specific amino-acid residues or specific exons (see Figure 2). By contrast, mutations related to striated muscle (68 % are missense mutations) are distributed all along the gene without clear relationships or hot spot for the various cardiac/skeletal clinical entities. Moreover, the three cardiac/skeletal clinical entities (EDMD, DCM-CD, LGMD1B) may co-exist within a same family.17,18 Interestingly, in line with previous reports,19 UMD-LMNA database analysis of patients with cardiac phenotype revealed that 33 % of the patients presenting isolated cardiac disease carry mutations leading to truncated proteins (nonsense, out-of-frame ins/del, splice-site) whereas only 8 % of the patients with cardiac and skeletal defects carry this type of mutations.
Cardiac Manifestations of Laminopathies
Cardiac Clinical Entities
Apart from the three main diseases initially described with LMNA mutation and cardiac expression, Emery-Dreifuss muscular dystrophy (EDMD), dilated cardiomyopathy and conduction defect (DCM-CD), limb-girdle muscular dystrophytype 1B (LGMD1B), additional entities were subsequently associated with the gene. These very few reports are related to DCM and early atrial fibrillation,20,21 left apical aneurysm without conduction defect,22 left ventricular non compaction,23 DCM and a quadriceps cardiomyopathy,24 inherited form of early onset myocardial fibrosis25 and arrhythmogenic right ventricular cardiomyopathy.26 Overlapping phenotypes have been also described as well as distinct phenotypes within a given family,17,18,21,24,27–29 so that it might be more appropriate to consider the global cardiac expression of laminopathies with its main features (dilated cardiomyopathy, conduction defect and ventricular arrhythmia), with or without skeletal muscle involvement.
Inheritance and Prevalence of LMNA Mutations
Mode of inheritance of cardiac laminopathies is autosomal dominant (50 % risk of transmission to offspring). Penetrance, or percentage of cardiac expression in mutation carriers, is not fully evaluated but appears very high and was estimated to be 100 % by the age of 60 years in one study.30 Exceptional studies reported on homozygous patients or digenism, associated with very early and severe phenotype.31–34 LMNA is one of the most frequent genes involved in dilated cardiomyopathy. In the largest study of 324 patients with DCM, prevalence of LMNA mutation was 7.5 % in familial cases and 3.6 % in sporadic cases, although the significance of some variants was unclear (in the absence of segregation in the family).35 No clinical criteria can distinguish DCM related to LMNA gene from other genetic or non genetic causes. However some clinical predictors of LMNA mutation were suggested in DCM patients: presence of skeletal muscle involvement, supraventricular arrhythmia, conduction defect, mildly dilated left ventricle, regardless of family history.36,37 Indeed, prevalence of LMNA mutations was increased to ~30 % in patients with DCM and conduction defect,37,38 but was very rare in patients with isolated DCM21,37 or isolated atrial fibrillation.39 Serum creatine kinase is increased in only part of mutation carriers (<30 %), with mild elevation (usually two-fold normal value) and is not considered as a strong predictor of LMNA mutation.12,17,36,37
Cardiac Complications
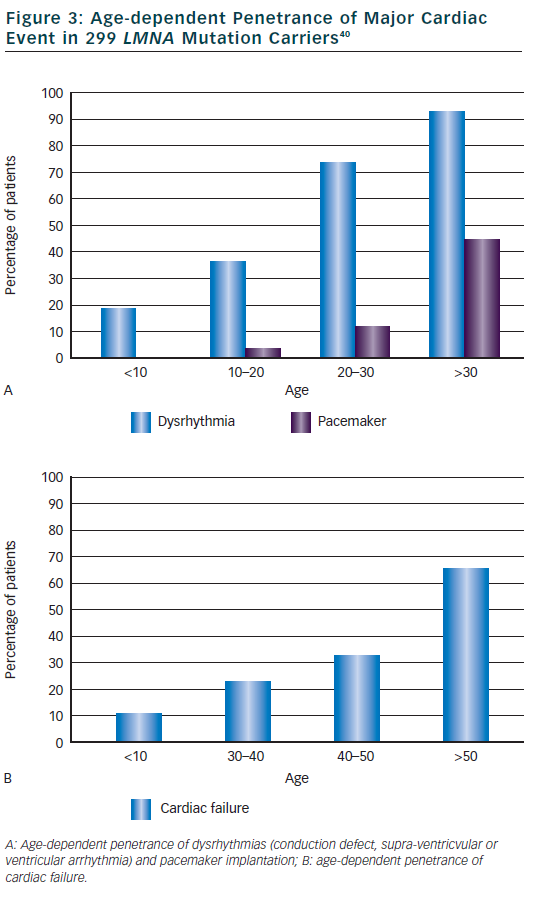
The main cardiac features were described in the princeps study about DCM and conduction defect related to LMNA mutations.12 Out of 39 patients with cardiac disease (mean age at onset 38 years, range 19 to 53) from five families, 34 patients (87 %) had atrioventricular block (AVC) or sinus node dysfunction, 23 (59 %) had atrial fibrillation or flutter and 25 (64 %) had DCM (heart transplant in six). Of note, 21 patients (54 %) were implanted with pace maker because of significant conduction defect. In this study, 20 additional relatives were mutation carriers but without cardiac abnormalities, all were younger than 30 years. Clinical features were then compiled in a meta-analysis of 299 mutations carriers from families with DCM, EDMD or LGMD1B.40 Dysrhythmia (conduction defect, supra-ventricular or ventricular arrhythmia) occurred early in life (2 children <10 years) and were highly penetrant: 74 % in those aged 20 to 30 years then 92 % of patients aged over 30 years (see Figure 3). Typical initial ECG shows a low P wave amplitude, PR interval prolongation and a normal QRS duration. Pace maker was implanted in 3 % of patients at age 10 to 20 and then increased to 44 % of patients after 30 years of age. Heart failure was reported at a later age, in 10 % in patients <30 years of age and increased progressively to 64 % of patients over 50 years. Ventricular arrhythmia was suggested as quite frequent in this meta-analysis since nearly half of sudden deaths (16 patients or 46 %) occurred in patients with a pace maker.40 Subsequent small studies or case reports observed that ventricular arrhythmia or appropriate defibrillator therapy may occur before myocardial dysfunction/DCM41,42 and sometimes as the first cardiac manifestation before conduction defect.43,44 One particular LMNA mutation (c.908- 909delCT) was suggested to be associated with a rapid progression of conduction defect and early sudden death.27
Cardiac Death
Clinical course of laminopathies is characterised by a poor prognosis and a high rate of major cardiac events. In a cohort of 105 DCM patients, cumulated survival was significantly worse in LMNA mutation carriers as compared to non carriers.36 By the age of 45 years, 55 % of mutation carriers had cardiovascular death or heart transplant as compared to 11 % in non carriers (p=0.0001 for global cumulated survival comparison). In the meta-analysis of 299 LMNA mutation carriers, cardiac death was observed in 76 patients (mean age 46 years) and sudden death was more prevalent than heart failure death (46 % of cardiac deaths versus 12 % respectively).40 Interestingly sudden death was similar in patients with isolated cardiac phenotype and in patients with cardiac and skeletal muscle phenotype.40 Recently, we reported a multicentre study that carefully examined the prognosis of 269 European LMNA mutation carriers during a median follow-up (FU) of 43 months.45 This retrospective study confirmed the high risk of ventricular arrhythmia: 18 % of patients developed sudden death, resuscitation or appropriate defibrillator therapy. In patients with defibrillator implanted at baseline, rate of appropriate therapy during FU was 13 % per year in secondary prevention and 8 % per year in primary prevention. Altogether, cardiac death due to sudden death was however lower than cardiac death due to heart failure (31 versus 47 % respectively).45
Prediction of Cardiac Death
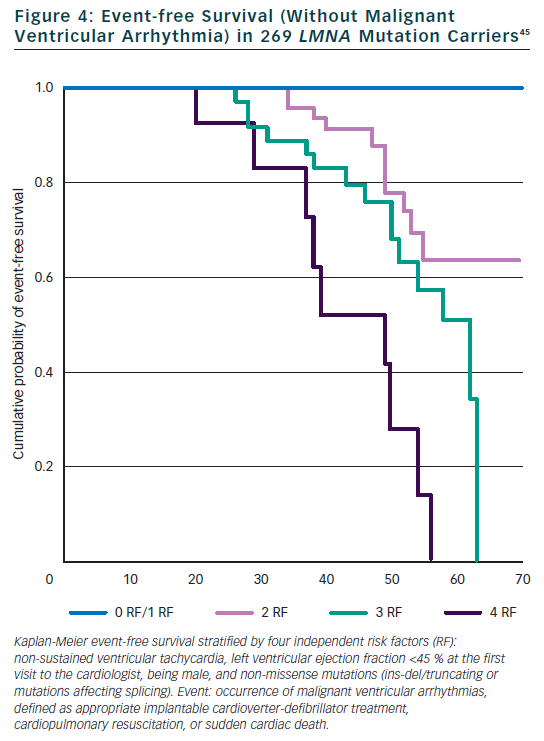
Few studies examined the predictive role of cardiac and non-cardiac features on the prognosis of mutation carriers. Our monocentric study of 94 Italian mutation carriers during a FU of 57 months identified two independent risk factors for total cardiac events: NHYA class III to IV and highly dynamic competitive sports for ≥10 years.30 In the same study, the type of mutation within the LMNA gene (splice site mutations) and history of competitive sports were independent risk factors for sudden cardiac death (SCD). In a study of 19 mutation carriers with conduction defect requiring a pace maker, but normal left ventricle ejection fraction (LVEF), cardioverter defibrillator was implanted systematically and eight patients (42 %) received appropriate defibrillator therapy during a FU of 34 months, suggesting defibrillator implantation in the presence of significant conduction defect.42 However significant conduction defect was not found to be a risk factor in two larger cohorts of 94 and 269 patients.30,45 Electrophysiological examination and inducible ventricular tachycardia was not a risk factor for SCD in the study of 19 patients.42 Recently, the study of 269 mutation carriers specifically focussed on the prediction of SCD.45 We identified four independent and cumulative risk factors for malignant ventricular arrhythmia (SCD, resuscitation, appropriate defibrillator therapy) (see Figure 4): LVEF <45 % at the first clinical contact, non-sustained ventricular tachycardia, male gender and a specific mechanism of LMNA mutation (non missense mutation: ins-del/truncating or mutations affecting splicing). No malignant ventricular arrhythmia occurred in patients with zero or one risk factor so that defibrillator implantation is suggested in the presence of at least two of these risk factors.45
Pathophysiology
Understanding of how mutations in a gene, LMNA, encoding ubiquitously expressed proteins, the lamin A/C, could give rise to defects specifically affecting the myocardium, remain so far an unsolved challenge. Numerous studies have tried to solve this mechanistic puzzle using tissues and/or cells available from mutated patients or various cellular and animal models that have been created in an attempt to phenocopy the human disease context.46
Lamin A/C expression explored in various explanted heart tissues from patients carrying nonsense LMNA mutations revealed reduced lamin A/C level, i.e. haplo-insufficiency in the cardiomyocyte nuclei.17,37 This is due to either the degradation of the mutated mRNA carrying a premature stop codon via the non sense mediated decay pathway or to the degradation of the corresponding truncated lamin A/C by the proteasome.47 As for missense mutations, the lamin A/C expression levels may vary from normal to reduced, underlying in those cases dominant negative effect of the mutant protein.37 Recently, we analyzed the quantitative gene expression of LMNA in 311 patients with DCM and observed that LMNA mRNA expression from peripheral blood, and myocardium, was reduced in DCM patients with LMNA mutation (p<0.001).48 Therefore, reduced mRNA expression level in blood might be a novel potential biomarker predictive for cardiac laminopathies. Ultrastructure analyses of explanted hearts carrying different mutations revealed focal disruptions and bleb formation of the cardiomyocyte nuclear membranes.37 These structural nuclear abnormalities are associated with altered chromosomal positioning and abnormal gene expression.49 The pathogenicity of several missense LMNA variants has been explored and desmonstrated abnormal intranuclear aggregates when overexpressed in heterologous context such as Hela cells50 or C2C12 myoblasts.39
To go further into the pathophysiological mechanisms, numerous murine models have been created.46 Mice lacking lamin A/C (Lmna-/-) display muscular dystrophy, dilated cardiomyopathy, signs of axonal neuropathy, reduction of adipose tissue and die by eight weeks of age.51–53 The heterozygous Lmna+/- mice, expressing 50 % of lamin A/C, develop arrhythymias and conduction defects by 10 weeks of age, in link with apoptosis of the conduction tissue, develop mild ventricle dilation by one year of age and 20 % of them die after eight months of age.6,54 Investigation of mechanotransduction pathways revealed abnormal desmin network and defective force transmission
in both Lmna-/- and Lmna+/- mice, associated in the latter with reduced hypertophic response after transaortic contstriction.52,55 The KI-LmnaH222P, knock-in mice reproducing the LMNA p.His222Pro mutation identified in EDMD patients, developed muscular dystrophy and DCM-CD similar to the human condition,56 at the homozygous state. Microarray analysis of cardiac gene expression performed at the incipience of the cardiac defects, desmonstrated abnormal activation of various branchs of the MAP kinase cascade (ERK1/2, JNK, p38α) and of AKT signalling pathway, linking the mutation to contractile dysfunction and myocardial fibrosis.57–59 Another knock-in model was created to study DCM-CD, the KI-LmnaN195K, reproducing a DCM-CD related LMNA mutation. Homozygous mice developed DCM-CD with minimal or no muscular dystrophy and died at three months due to arrhythmia. The transcription factor Hf1b/Sp4 and the connexin 40 and 43 were misexpressed/mislocalised in mutant hearts. As for knock-out mice, desmin displayed abnormal organisation.60
Altogether, these observations suggest that LMNA mutations may cause cardiomyopathy by haploinsufficiency and/or dominant negative effect, with disrupting the internal organisation of the cardiomyocyte and/or altering the gene expression of transciptional factors and proteins involved in various signalling pathways, all essential to normal cardiac development, ageing and function.
Perspective for Therapeutics
Even if the pathophysiology of cardiac laminopathies remains to be fully elucidated, the analyses of the mouse models that phenocopy the human disease allowed to test several therapeutical approaches. The first approach tested has been pharmacological. Inhibitors of different branchs of the MAPK and AKT pathways in the KI-LmnaH222P mice not only delay the onset but also slow down the progression of the cardiac disease.57,61–64 As some MAPK inhibitors have been tested in humans during clinical trials for other indications, their efficacy and safety in laminopathy deserved further investigations. Calcium sensitiser also gives beneficial effects on the contractile function and lead to increased survival of the mice.65
Another possible approach is gene therapy. Indeed, combination of stem cell-based approaches with gene-editing technologies has been recently reported for laminopathy and represents an attractive therapeutical strategy.66 Homologous recombination-based gene correction of multiple LMNA mutations using helper-dependent adenoviral vectors (HDAdVs) demonstrated a highly efficient and safe method for correcting mutations in human induced pluripotent stem cells (hIPSC).66 The fast advancing field of hIPSC67 will certainly lead to innovative therapeutic approaches in the near future.
Finally, improvement in therapeutic management might come from very early treatment with drugs already used in human in heart failure. We designed the Pre-clinical mutation carriers from families with dilated cardiomyopathy and ACE inhibitors (PRECARDIA) trial that is a multicentre randomised double blind trial (perindopril versus placebo) proposed to participants without significant systolic dysfunction but who are mutation carriers from families with dilated cardiomyopathy (whatever the underlying gene) in order to delay or prevent the systolic dysfunction. The enrollment of participants is underway.68
Key Messages for the Cardiologist
Identification of mutations in LMNA gene in clinical practice is rapidly increasing so that cardiologists are more and more frequently faced to difficult questions regarding optimal management of patients and relatives. The following section briefly summarises the management we propose, that reflects our personal view and is based on available data and our experience.
When Should the Cardiologist Suspect a Laminopathy?
The diagnosis should be suspected in a patient with DCM and conduction defect (atrioventricular block or sinus node dysfunction) or DCM and skeletal muscle abnormality (muscle weakness/wasting, tendon contractures, increased creatine kinase level) or DCM preceded (few years before) by surpa-ventricular or ventricular arrhythmia, whatever the familial context (familial or sporadic form).
How Can we Confirm a Laminopathy?
The diagnosis, when suspected because of the above criteria, should be confirmed by genetic testing with the analysis of the LMNA gene. When conventional LMNA direct sequence analysis does not identify a mutation in suggestive pedigrees, alternative strategies such as multiplex ligation-dependent probe amplification might be discussed to detect large gene rearrangements.69 Differential diagnoses include other genes such as SCN5A, desmin, DMPK (Steinert), dystrophin and desmosomal genes.
What is the Clinical Course of a Laminopathy?
The disease is associated with a poor prognosis, related to heart failure and to sudden cardiac death (caused by conduction defect or ventricular arrhythmia). First cardiac expression includes conduction defect (AVB type 1) or supra-ventricular arrhythmia, ususally between 20 and 30 years of age. DCM is common between 30 and 50 years of age. Ventricular arrhythmia can occur at various stage of the disease. Skeletal muscle weakness or wasting can be absent or can occur at a late stage. A subject can develop only part of the features and the cardiac expression or chronology may be different among relatives within a given family.
What is the Screening that can be Proposed to the Patients and the Relatives?
The diagnosis in a patient should lead to regular cardiac examination including electrocardiogram (ECG), echocardiography, Holter-ECG and exercise testing. Skeletal muscle examination and creatine kinase dosage are also warranted. Cardiac examination, or predictive genetic testing, is proposed to all first degree-relatives within the family (from 10–12 years of age).70
What is the Therapeutic that should Currently be Proposed?
Competitive sport should be discouraged in all LMNA mutation carriers, whatever the stage of the disease and the presence of cardiac abnormalities. Myocardial systolic dysfunction, supra-ventricular arrhythmia and conduction defect should be regularly screened for and lead to non-specific management (no specific indication for pace maker implantation for example) except careful use of drugs with negative chronotropic effect in the absence of a pace maker. In contrast, there is a high and early risk of ventricular arrhythmia. Cardiovertor defibrillator can be proposed in the presence of two criteria among the four following: LVEF <45 %, non sustained ventricular tachycardia, male gender and non-missense LMNA mutation. If only male gender and non-missense LMNA mutation are present, the situation is however not sufficient to propose a defibrillator. Although based on lesser evidence, it seems also possible to propose defibrillator implantation in the presence of significant conduction defect that requires a pace maker implantation.