









Sarcoidosis is an inflammatory granulomatous disease that can affect any organ. Systemic sarcoidosis is known to affect young adults, with a second peak in women >50 years of age, as demonstrated in Scandinavian and Japanese studies.1–4 In the US, the lifetime risk of sarcoidosis is 2.4% for black people and 0.85% for white people.1 The incidence of cardiac involvement has been increasingly recognised, with one large 25-year Finnish cohort study reporting an exponential increase from 1988 to 2012, with a prevalence of 2.2 per 100,000 adults.2 Among patients with systemic sarcoidosis, an estimated 5% will have clinically manifest cardiac sarcoidosis (CS), whereas more than 25% may have evidence of cardiac involvement on autopsy or imaging studies.1,5
The diagnosis of CS can be challenging given the low sensitivity of endomyocardial biopsy. However, advanced cardiac imaging techniques permit non-invasive detection of cardiac involvement. Accordingly, current guidelines provide both histological and clinical pathways for diagnosis, and emphasise the important role of cardiac imaging.6 The diagnosis of cardiac involvement in sarcoidosis has important clinical and prognostic ramifications, including an increased risk of heart failure, ventricular arrhythmias (VAs) and sudden death.
Optimal management strategies of patients with CS are evolving as the evidence base expands. Immunosuppression remains the mainstay of therapy, and corticosteroids are often the initial treatment of choice. However, steroid-sparing agents have emerged as an important adjunctive treatment in an effort to decrease the long-term side effects related to corticosteroid therapy. Furthermore, many studies have refined our understanding of which patients are at increased risk for developing VAs and may benefit from device-based therapy.7
In this review, we provide an update on the diagnostic criteria for CS, discuss the utility of imaging modalities in the diagnosis and monitoring of CS and review current management strategies for the cardinal clinical manifestations of CS, namely conduction disease, arrhythmia and cardiomyopathy.
Diagnosis of Cardiac Sarcoidosis
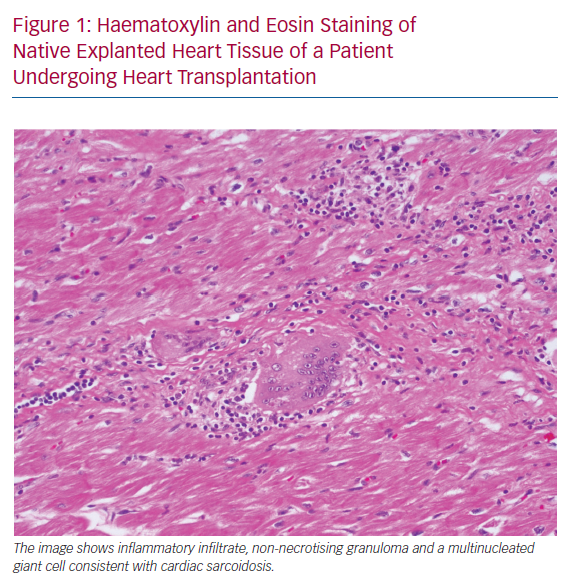
Despite multiple existing guidelines and diagnostic criteria for CS, the largest current limitation is the emphasis on a tissue diagnosis. Histopathological examination of the myocardium involved by sarcoidosis reveals non-caseating granulomas, multinucleated giant cells and asteroid bodies (Figure 1). Eosinophils and myocyte necrosis are rare and can help distinguish CS from other causes of inflammation, such as giant cell myocarditis. In certain presentations of cardiomyopathy, endomyocardial biopsy is indicated for diagnosis, but the role of biopsy is limited in CS due to low sensitivity, which may be due to the patchy nature of the disease.8 In cases where biopsy is pursued, guidance with electroanatomical voltage mapping may increase the diagnostic yield.9 In patients with a clinical diagnosis of CS, a positive endomyocardial biopsy is known to be a poor prognostic indicator.10
One of the initial guidelines for the diagnosis of CS was developed by the Japanese Ministry of Health and Welfare (JMHW) in 1993 and later revised in 2007.11,12 These guidelines included characteristic clinical manifestations as major criteria and late gadolinium enhancement cardiac MRI (LGE-CMR) and perfusion defect on nuclear imaging as minor criteria. However, these guidelines did not include abnormal PET imaging as a criterion.11,12 Furthermore, it has been suggested that advanced imaging techniques may have a higher sensitivity for CS diagnosis compared with the modified JMHW criteria.13,14
The World Association of Sarcoidosis and Other Granulomatous Disorders (WASOG) provided an alternative approach to diagnosis based on the results of a detailed survey completed by sarcoidosis experts.15,16 The WASOG Sarcoidosis Organ Assessment Instrument established whether specific pathological, laboratory, clinical and imaging criteria supported a highly probable, probable or possible diagnosis of CS, where the experts voted using Delphi study methodology and consensus was achieved with ≥70% agreement.15 In this diagnostic approach, a ≥90% likelihood of CS matched a highly probable diagnosis of CS, a ≥50% likelihood matched a probable diagnosis and a <50% likelihood matched a possible diagnosis.
The Heart Rhythm Society (HRS) published an expert consensus statement in association with the American College of Chest Physicians, American Heart Association, Asia Pacific Heart Rhythm Society, European Heart Rhythm Association and WASOG in 2014.6 The HRS guidelines recognised both histological (definite) and clinical (probable) pathways for the diagnosis of CS. Importantly, these guidelines included abnormal PET or CT as a diagnostic criterion. However, both abnormal PET/CT and abnormal LGE-CMR were still considered minor, rather than major, criteria. This made the diagnosis of isolated CS challenging in cases where endomyocardial biopsy was not feasible or was negative.
Not until recently has there been a movement to diagnose CS without histopathological confirmation. These efforts were supported by the recently published Japanese Circulation Society guidelines, which include a clinical diagnosis pathway using abnormal PET or CT and LGE-CMR as major criteria for CS diagnosis.17 Furthermore, these guidelines outline a specific pathway for the clinical diagnosis of isolated CS in cases where endomyocardial biopsy is not available.17 This recent shift in diagnostic practice will have future implications on disease definition when deciding on treatment strategies or inclusion in research studies.
Imaging
Various imaging modalities play a role in the diagnosis and monitoring of cardiac involvement in sarcoidosis. In addition to more traditional echocardiography and MRI, the greatest advancement has been in nuclear imaging, with a shift away from gallium scans to the use of cardiac PET.
Echocardiography
Although echocardiography has limited sensitivity and specificity for the diagnosis of CS, it is often the initial imaging study acquired in the evaluation of patients with suspected cardiomyopathy. Echocardiographic findings that may support the diagnosis of CS include ventricular hypertrophy, diastolic dysfunction or restrictive filling pattern, and systolic dysfunction of either the left ventricle (LV) or right ventricle with non-coronary distribution wall motion abnormalities and aneurysms.18,19 The more recently developed speckle tracking echocardiography has allowed measurement of global longitudinal strain (GLS) in CS. As in other cardiomyopathies, GLS may have both diagnostic and prognostic utility in CS, and is independently associated with poorer clinical outcomes in patients with CS.20,21
Cardiac PET
18F-Fluorodeoxyglucose (FDG) is a glucose analogue taken up by macrophages.22 Cardiac PET using 18F-FDG has emerged as a cornerstone in the clinical diagnosis, prognostic evaluation and monitoring of therapy in patients with CS. Several patterns of 18F-FDG uptake have been described in CS, namely focal uptake and focal-on-diffuse uptake.23,24 Diffuse uptake is often interpreted to represent poor suppression of normal myocardial glucose uptake. Metabolic imaging is often performed in conjunction with perfusion imaging. In these cases, the classic pattern demonstrated in CS is one of ‘perfusion–metabolism’ mismatch, in which areas of 18F-FDG uptake correspond to areas of reduced or absent perfusion.23 FDG-PET/CT has a fair diagnostic accuracy for CS, with a recent meta-analysis reporting a pooled sensitivity of 89% and specificity of 78%.25 FDG-PET/CT may also be complementary to LGE-CMR in the diagnosis of CS.24 Abnormal FDG uptake is also important for prognosis and is associated with increased rates of VAs and death, especially when located in the right ventricle.14 Finally, serial PET imaging is useful in monitoring disease activity and response to immunosuppressive therapy.23,26
Cardiac MRI
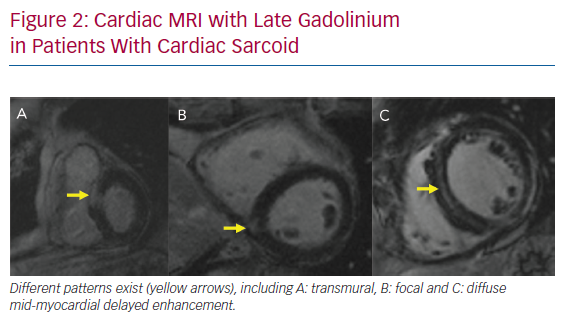
LGE-CMR plays an important role in the diagnosis of CS and risk stratification of patients with CS. The main strength of LGE-CMR is its early detection and high sensitivity.27 Patel et al. demonstrated a higher sensitivity of LGE-CMR that that of the JMHW criteria.13 Although the presence of LGE may be a non-specific finding in the evaluation of non-ischemic cardiomyopathy, multifocal distribution, high signal intensity and contiguous extension from the left to the right ventricle may increase the specificity of this finding for the diagnosis of CS (Figure 2).24 Many studies have also demonstrated the prognostic utility of LGE-CMR in patients with CS.28–30 A meta-analysis by Coleman et al. including 760 patients with known or suspected CS demonstrated that the presence of LGE is associated with an odds ratio of 10 for the composite endpoint of VAs and all-cause mortality.31 Hybrid CMR-PET imaging has also been proposed as a future tool in CS, because studies have shown incremental value to this approach in determining disease activity and pattern.32
Clinical Manifestations
When involving the heart, sarcoidosis classically presents with atrioventricular (AV) conduction disease, arrhythmia and cardiomyopathy causing heart failure. Less commonly, CS may manifest as pericardial, valvular or coronary disease.
Heart Block
AV nodal disease is a common mode of presentation among patients with CS. In a series of 110 Finnish patients with histologically confirmed CS, 48 (45%) presented with AV nodal disease, 35 (32%) of whom had third-degree AV block requiring permanent pacemaker implantation.2 Among patients with CS who present with other initial clinical manifestations, there are no known predictors for the development of AV nodal disease; however, LGE in the basal anteroseptal region on CMR may portend increased risk for AV nodal disease.33 Treatment includes corticosteroids and device therapy, both of which are discussed below.
Atrial Arrhythmias
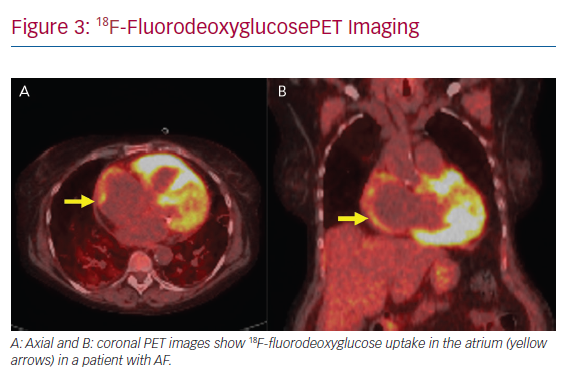
Atrial arrhythmias are common in CS. Hypotheses as to the mechanism of atrial arrhythmias include triggered activity from active inflammation to re-entry secondary to scar formation (Figure 3). In a study of 100 patients with CS, supraventricular arrhythmias were detected in 32% based on ambulatory ECG and cardiovascular implantable electronic device monitoring, with the most prevalent atrial arrhythmia being AF in 18% of all patients studied.34 Cain et al. performed a CMR study in which 36% of patients with ventricular myocardial LGE had documented atrial arrhythmias.35 CS is known to infiltrate the atrium, based on a 1977 clinicopathological study in which five of the 26 hearts studied had sarcoidosis granulomas in the right or left atrium.36 In that series, four had documented atrial arrhythmias.36 In a similar study performed by Tavora et al., the prevalence of atrial involvement of granulomas was lower at 3.7%.37
Treatment of atrial arrhythmias in the setting of CS has been limited to case reports. Although immunosuppression remains the cornerstone for treatment of inflammation, to date there are no specific guidelines on the role of anti-arrhythmic therapy and catheter ablation in patients with CS. It is generally agreed that class I anti-arrhythmics should be avoided, whereas beta-blockers, calcium channel blockers and drugs that block potassium currents (e.g. sotalol, dofetilide and amiodarone) are acceptable choices.6
Ventricular Arrhythmias
Patients with CS are at increased risk of ventricular tachycardia (VT) and sudden cardiac death (SCD), although the precise incidence of VAs in CS is not well defined.7 Kandolin et al. observed that, among 18 patients presenting with AV nodal disease and ultimately diagnosed with CS, 10 went on to develop VA during a mean follow-up of 48 months.38 In a much larger study, Nordenswan et al. observed that among 143 patients with CS and Mobitz II second-degree heart block or complete heart block, 42 developed VT or SCD during a median follow-up of 2.8 years.39 Importantly, even patients with CS and preserved LV systolic function are at increased risk of VA.14,39 Among the 90 patients with preserved LV ejection fraction (LVEF) in the study of Nordenswan et al., the 5-year incidence of subsequent SCD or VT was 24%.39
Although VT is generally monomorphic in CS, polymorphic VT has also been described.40 Myocardial scar resulting from granulomatous inflammation is thought to be the dominant substrate for VT in patients with CS; however, the role of active inflammation in arrhythmogenesis has not been well characterised and may be an important therapeutic target in patients presenting with VT.41–43 Circuits supporting re-entrant VT may localise to either ventricle, and to any myocardial depth (i.e. subepicardial, mid-myocardial, subendocardial or transmural).41 Finally, the His–Purkinje system may be an important component of the arrhythmogenic substrate in some patients with CS.42
Risk stratification for SCD may be challenging in CS. Non-invasive strategies include LGE on CMR and abnormal 18F-FDG uptake on cardiac PET. As mentioned above, in a meta-analysis including 10 studies and 760 patients with CS undergoing CMR, those with LGE on CMR had a 10-fold increased risk of the combined endpoint of VA or all-cause mortality over a mean follow-up of 3 years.31 Among those with LVEF >50%, the presence of LGE conferred a 19-fold increased risk of the combined endpoint.31 Blankstein et al. studied 118 patients with suspected CS, among whom the presence of abnormal 18F-FDG uptake corresponded to an approximately fourfold increased risk of VT or death over a median follow-up of 1.5 years.14
The role of invasive risk stratification with programmed electrical stimulation (PES) has been assessed in several analyses (Figure 4). Among 25 patients with CS undergoing PES at the Johns Hopkins University School of Medicine, 10 had inducible VA, all of whom had clinical VA events during a mean follow-up of 5 years, whereas 15 had no inducible VA, only one of whom went on to have clinical VA.44 In a larger study, Mehta et al. observed that six of the eight patients who had inducible VA on PES had clinical VA events over a mean follow-up of 5 years, whereas only one of the 69 who had no inducible VA went on to have clinical VA.45 Electrophysiological studies for the purpose of arrhythmic risk stratification in patients with CS and LVEF >35% is a Class IIb recommendation in the current guidelines.6
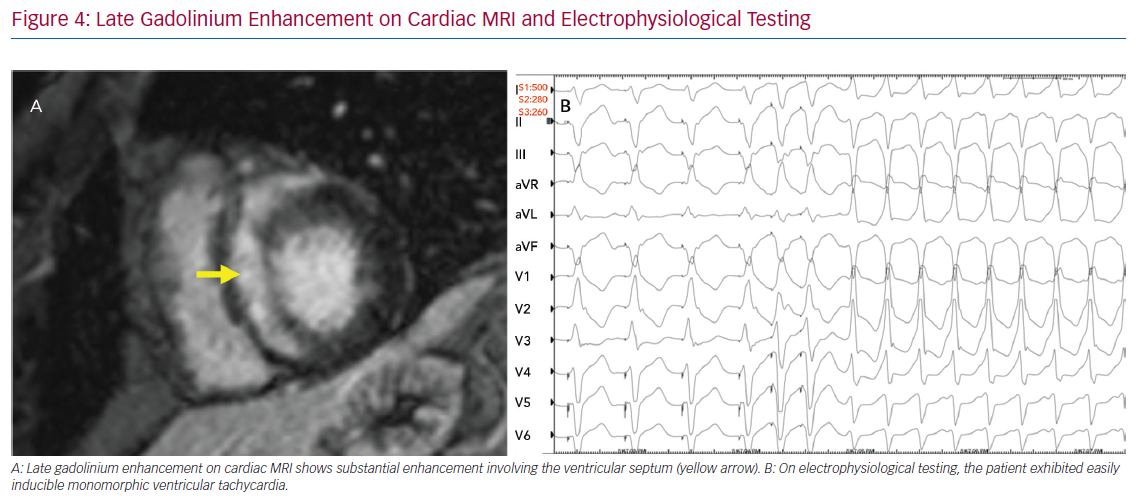
The treatment of VA in CS includes medical therapy in the form of both anti-arrhythmic drugs and immunosuppression, device therapy in the form of secondary prevention ICD and catheter ablation. These are discussed separately below in the ‘Management’ section.
Heart Failure
Sarcoidosis is gaining increasing recognition as an aetiology for non-ischemic cardiomyopathy, particularly with advancements in cardiac imaging. Depending on the cohort studies, approximately 50% of patients with CS have cardiomyopathy.46 Granulomatous inflammation and subsequent scarring can result in both systolic ventricular dysfunction and diastolic dysfunction and a restrictive physiology similar to other infiltrative cardiomyopathies. Sarcoidosis can also involve either ventricle, and may be a cause for isolated right ventricular dysfunction.47 CS requires distinction from other cardiomyopathies, which can have overlap in presentation. For example, arrhythmogenic right ventricular cardiomyopathy (ARVC) also presents in relatively young patients with VA and right ventricular dysfunction, and patients with sarcoidosis may meet Task Force Criteria for ARVC.48 However, management is significantly different for ARVC and CS.
Patients with CS can also present like patients with giant cell myocarditis, a lethal form of myocarditis characterised by acute cardiac failure, VA and conduction disease that is diagnosed on endomyocardial biopsy and treated with immunosuppressive therapy, but often requires mechanical support and heart transplantation. Compared with patients with dilated cardiomyopathy, patients with CS-related cardiomyopathy have been noted to more likely be women and have AV block, LV hypertrophy and focal LV wall involvement.49 These patients have poorer prognosis than those with other dilated cardiomyopathies.49 In addition, when manifesting with isolated CS, patients are more likely to present with LV systolic dysfunction than patients who also have extracardiac disease. In the cohort of Kandolin et al., 69% of those with isolated CS had LV systolic dysfunction on presentation, compared with 41% of those with both extracardiac and CS.39
The presence of heart failure has implications on survival among patients with CS, as demonstrated in several retrospective cohorts. In the Finnish cohort, 10-year transplantation-free cardiac survival was only 53% among those presenting with heart failure, whereas the overall cohort survival was 83%.1 In a more recent multicentre analysis, Fussner et al. described a cohort of 91 patients with CS, of which 47 (52%) had a primary presentation of cardiomyopathy.46 Those with cardiomyopathy had a significantly lower survival free of LV assist device (LVAD) placement, heart transplantation or death.46 Among patients with sarcoidosis-related cardiomyopathy, independent predictors of mortality include worse New York Heart Association functional class, larger LV diastolic dimension, lower LVEF and the concomitant presence of sustained VT.39,50 In addition, right ventricular involvement, particularly on FDG-PET scans, is predictive of poorer survival.14
Management
Immunosuppression
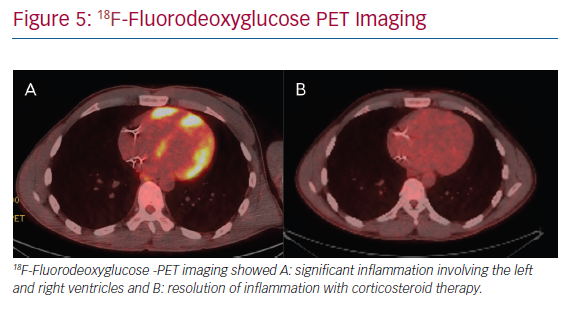
The mainstay of medical therapy for CS, as with other organ involvement, is immunosuppression, namely in the form of corticosteroids (Figure 5). In addition, a number of different steroid-sparing agents may be used to avoid untoward side-effects of chronic corticosteroid use. Data on immunosuppressive management of CS is largely extrapolated from non-cardiac sarcoidosis literature and from limited retrospective cohorts of CS patients.51 Consensus guidelines and prospective randomised studies are lacking. Therefore, ambiguity and clinical practice variation exist in the treatment of CS.
Immunosuppressive regimens are generally tailored towards response to treatment, assessed both by clinical events and imaging. In an attempt to further elucidate the role and efficacy of corticosteroids in CS, Sadek et al. performed a meta-analysis of 10 studies comprising a total of 257 patients with CS who received corticosteroids and 42 who did not.51 That meta-analysis was limited by fair-quality studies that were mostly small, single-centre retrospective cohorts, and thus limited any significant conclusions regarding the efficacy of corticosteroids. Although randomised data are lacking, corticosteroid therapy is thought to play an important role in the treatment of AV nodal disease in CS. Among the 35 patients with third-degree AV block in the series of Kandolin et al., seven recovered AV conduction after the initiation of corticosteroids.2 In the meta-analysis of 10 studies assessing the utility of corticosteroids in CS by Sadek et al., 27 of 57 patients with AV nodal disease treated with corticosteroids showed clinical improvement, whereas none of the 16 patients with AV nodal disease not treated with corticosteroids showed clinical improvement.51
Observational data support the use of corticosteroids in the treatment of VA in patients with CS and evidence of active inflammation.43 In that study, Yalagudri et al. studied 18 patients presenting with VT who were ultimately diagnosed with CS. All patients underwent FDG-PET examination, with 14 demonstrating abnormal myocardial FDG uptake. Among these, nine were successfully treated with a combination of prednisolone and methotrexate and did not require chronic maintenance with antiarrhythmic drugs, whereas five required either intensification of immunosuppression or radiofrequency ablation.43
The data on LVEF responses to prednisone are mixed, with some studies suggesting those with severely depressed LVEF tend to improve more and others reporting the opposite.2,46,51 However, the efficacy of steroids in suppression of FDG uptake, and the resulting association with clinical improvement, has been demonstrated in small series.52–54
The dosing and duration of prednisone treatment for CS varies widely. In a retrospective analysis by Yazaki et al. of 95 Japanese patients, 75 of whom received prednisone, survival was similar among those who received ≤30 and >30 mg prednisone.50 Our group recently described a cohort of 32 patients with CS undergoing serial FDG-PET and treatment with corticosteroids.53 There was a significant reduction in cardiac inflammation measured by maximum standard uptake value and the number of LV segments involved after steroid treatment, but results were similar for patients who received high (≥40 mg) and low (<40 mg) doses of prednisone upfront.53
In the context of limited available data, the general approach to treatment of CS includes early initiation of prednisone at 30–40 mg/day, with subsequent monitoring and tapering as tolerated. If the decision to discontinue treatment is made, patients should be monitored closely due to the risk of clinical worsening after discontinuation.55 Steroid-sparing agents used most commonly include mycophenolate mofetil, methotrexate and azathioprine.46,52 Biological agents, such as tumour necrosis factor alpha inhibitors, may be reserved for refractory disease.56–58 Given the current limitations, there has been a call for randomised clinical trials to address gaps in knowledge regarding the treatment of CS. The Cardiac Sarcoidosis Multi-Center Randomized Controlled Trial (CHASM CS–RCT) is the first of its kind and is currently evaluating low- versus standard-dose prednisone in combination with methotrexate.59
Cardiac Medications
In addition to immunosuppression, patients with CS should be treated with guideline-directed medical therapies (GDMT) for electrophysiological and heart failure manifestations. In the setting of reduced LV systolic function, treatment with heart failure GDMT is typically initiated and includes beta-blockers and renin–angiotensin system blockade with angiotensin-converting enzyme inhibitors, angiotensin receptor blockers or a neprilysin inhibitor–angiotensin receptor combination (sacubitril/valsartan).60 For symptomatic patients, the addition of a mineralocorticoid receptor antagonist is indicated. Diuretics are used for symptomatic management of volume overload. Data are lacking on outcomes related to the use of heart failure GDMT specifically in CS; however, the benefit of such therapy is extrapolated from existing, well-established data in patients with reduced LVEF.61,62
Similarly, although often used as adjunctive therapy to ICDs and catheter ablation, there are limited and inconclusive data regarding the use of anti-arrhythmic drugs in the management of patients with CS and VA.51 Class I anti-arrhythmics should be avoided in the setting of myocardial scar and structural heart disease. Thus, class III anti-arrhythmics, such as sotalol, dofetilide and amiodarone, are preferred for the management of atrial and ventricular arrhythmias.
Device Therapy
Device therapy with permanent pacemakers and/or ICDs is an essential component of the therapeutic approach to patients with CS and arrhythmic events. In general, indications for pacemaker implantation among patients with CS mirror those applying to patients with bradyarrhythmias.6 Implantation of a permanent pacemaker is the definitive treatment for AV nodal disease in CS, and may be appropriate even in cases of transiently recovered AV conduction.
Among patients with CS presenting with a VA event, secondary prevention ICD implantation is warranted, whereas among patients with CS and LVEF <35% despite optimally tolerated GDMT, primary prevention ICD implantation is warranted. The utility of primary prevention devices among patients with CS and mid-range or preserved LVEF is less straightforward. Although PES for risk stratification is supported only with Class IIb guideline recommendations, in patients with inducible VA the placement of a primary prevention ICD carries a Class IIa recommendation.6 Finally, in patients with CS and AV nodal disease, there are Class IIa guidelines for implantation of a primary prevention ICD rather than a pacing system alone, even in patients with preserved LVEF.6 Indeed, available data support this recommendation and suggest a high rate of subsequent SCD among patients with CS who initially present with AV nodal disease, as discussed above.39 Among patients with depressed LVEF needing a high burden of pacing, or with heart failure with a left bundle branch block, chronic resynchronisation therapy is warranted and has been shown to be as efficacious in patients with CS as in patients with other non-ischemic cardiomyopathies.63
The risks associated with device implantation in patients with CS may exceed those in the broader population. Kron et al. studied 235 patients with CS and primary or secondary prevention ICDs.64 The overall rate of inappropriate tachytherapies was 24%. In all, 41 patients experienced 46 other adverse events, including seven device-related infections and 25 lead dislodgements or fractures.64 Although there is no definitive evidence to suggest a higher rate of device-related infections among patients with CS, given frequent concomitant treatment with immunosuppressive agents, heightened alertness for possible device-related infections may be reasonable. Indeed, in the study by Kron et al., among six patients with device-related infections, five were being treated with immunosuppression and two of the infections involved epicardial systems.64
Catheter Ablation
Depending on the substrate (i.e. scar or inflammation mediated), catheter ablation may be an effective component of the therapeutic approach in patients with CS and VA.7 The efficacy of catheter ablation in patients with CS and VA has not been assessed in a randomised manner, but is reported to range from 25% to 56% if complete absence of recurrent VA is the endpoint.41,43,65–69 For this reason, catheter ablation is recommended only in cases of VA refractory to antiarrhythmic drugs and immunosuppression, with Level IIa strength.6
Advanced Heart Failure Therapies
Despite immunosuppression and heart failure GDMT, a significant proportion of patients will not recover LV function or may have a decline in LVEF over time.46 In patients who develop refractory heart failure or VA, the primary drivers of mortality in this cohort, advanced heart failure therapies, such as mechanical circulatory support or heart transplantation, may be considered.
When evaluating patient candidacy for advanced heart failure therapies, there are a few special considerations specific to the CS population. The extent of extracardiac organ involvement should be thoroughly assessed to ensure longevity after heart transplantation and safety of undergoing cardiac surgery. With regard to LVAD evaluation, right ventricular involvement should be assessed by imaging and using guideline-directed haemodynamic assessments in order to determine the risk of right ventricular failure after LVAD placement.70 CS patients presenting primarily with refractory VA may benefit from a direct transplant approach, because LVAD placement may contribute to further scar and arrhythmic nidus formation. Notably, a subset of patients may go unrecognised and only attain a diagnosis of sarcoidosis after examination native heart tissue at the time of LVAD placement or transplantation.71,72
Analyses from the United Network for Organ Sharing (UNOS) have demonstrated similar or better post-transplant survival outcomes for patients undergoing transplant for CS compared with other cardiomyopathies.73,74 In addition, among those undergoing mechanical circulatory support as a bridge to transplantation, survival was similar in those with and without CS.74 There is some theoretical risk of sarcoidosis recurrence in the transplanted heart, but in published case reports this is typically in the setting of weaning off corticosteroids.75,76 More contemporary single-centre case series of cardiac transplantation in CS have reported no recurrence of CS in the allograft.72,77 After transplantation, the general approach is to maintain indefinite low-dose prednisone therapy in patients transplanted for CS. Patients undergoing LVAD placement may also be maintained with immunosuppression afterwards, weighing the benefits of sarcoidosis disease suppression with LVAD-related infection risk.
Cardiac Sarcoidosis in 2020: Where We Are and a Look to the Future
CS is an increasingly recognised cause of heart block, VA and cardiomyopathy. Past limitations in diagnosis and management have included small single-centre or single-country studies limiting generalisability, a need for histopathological diagnosis and a lack of prospective trials for treatment efficacy. Over the past decade, advancements in cardiac imaging and newer expert consensus guidelines have lifted some of the prior challenges to the diagnosis of CS. These advancements may allow earlier recognition, and thus treatment, of CS moving forward, ideally prior to the development of irreversible cardiac inflammation and fibrosis. Although corticosteroids are the mainstay of therapy, prospective clinical trials are needed to determine the optimal dosing and treatment duration. In addition, retrospective studies of steroid-sparing agents in CS are only now starting to be published. The role of these agents needs to be further defined in efforts to decrease the morbidity associated with corticosteroids.
Much remains to be learned on how best to diagnose and manage patients with CS. For example, how do we best screen patients for CS who have known extracardiac sarcoidosis? How do we better risk stratify patients with preserved or low normal LVEF with no prior history of VA? Should we treat patients with clinically silent CS? Ultimately, prospective multicentre studies are needed to elucidate answers to these questions to move the care of patients with CS forward.
Clinical Perspective
- The clinical manifestation of cardiac sarcoidosis (CS) includes advanced-degree heart block, atrial tachycardia, ventricular arrhythmias and heart failure.
- Cardiac MRI and PET imaging are important imaging tools for the diagnosis of CS and risk stratification.
- In selected patients, ICD therapy is warranted to decrease the risk of sudden cardiac death.
- Immunosuppressive therapy is the mainstay of treatment for active, inflammatory CS.