








Sudden unexplained death syndrome (SUDS)1 is rare in the young but when it occurs it is devastating for family and friends, and affects whole communities. That it can affect fit, athletic individuals and may be related to competitive sports only adds to the sense of incomprehension and injustice felt by wider society. In comparison with the older population, where sudden death is more common and mostly a consequence of ischaemic heart disease,2 in the young it is more commonly related to heritable cardiac disease such as cardiomyopathies, channelopathies and aortopathy. Therefore, systematic evaluation of family members of sudden death victims can reveal other affected individuals and potentially prevent further deaths.
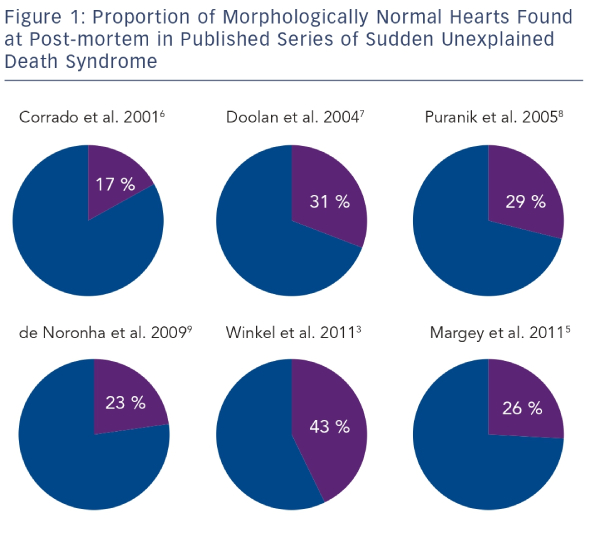
The reported incidence of SUDS is variable.3–5 This is due mainly to methodological differences between studies and differing population characteristics. Winkel et al.3 analysed the death certificates of all deaths occurring in persons aged less than 35 years in Denmark over a seven-year period. They identified 625 cases of SUD and calculated an incidence of 2.8 per 100,000 person-years. A similar study by Papadakis et al.4 conducted in England and Wales found a rate of 1.8 per 100,000 person-years. The rate of sudden death in persons aged 14–35 in the Republic of Ireland was reported as 2.9 per 100,000 person-years.5 Studies of post-mortem findings in cases of SUDS in the young have shown a high prevalence of a structurally normal heart.3,5–9 In the context of a normal post-mortem examination and negative toxicology, SUDS is referred to as the sudden arrhythmic death syndrome (SADS).1 SADS has been shown to represent 17–43 % of all sudden cardiac deaths in the young (see Figure 1).
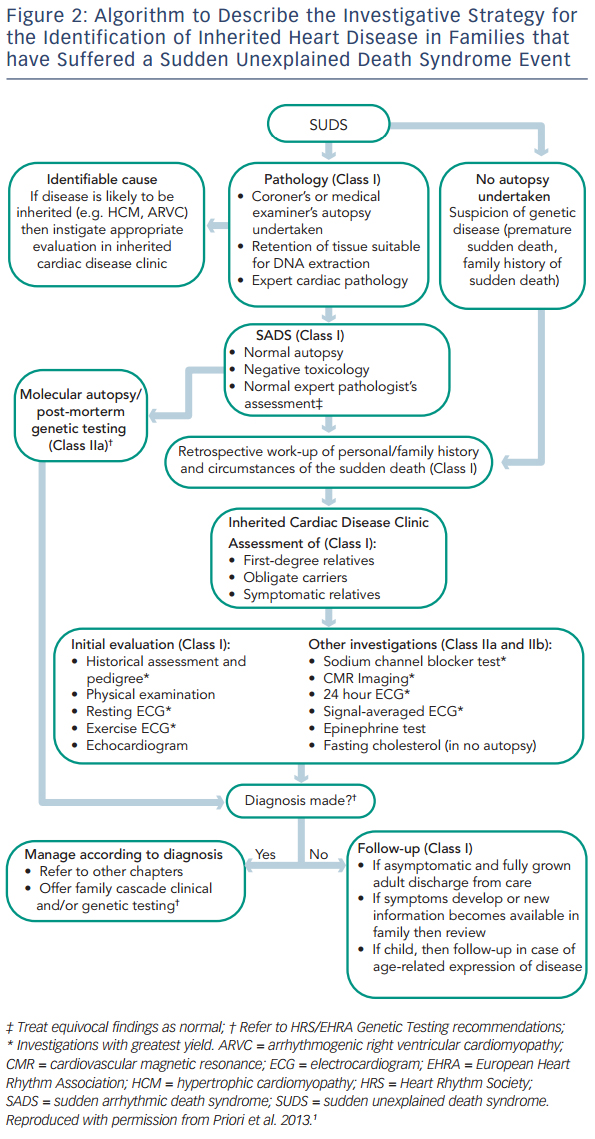
The European Heart Rhythm Association (EHRA), Heart Rhythm Society (HRS) and Asia Pacific Heart Rhythm Society (APHRS) recently published an expert consensus statement on the management of inherited arrhythmia syndromes.1 This outlines the need for investigation of family members of SUDS victims (see Figure 2). Systematic investigation of first-degree relatives is recommended, with subsequent focused investigation of further relatives in families with positive findings. The recommendation is based upon previous studies that have shown that familial assessment reveals affected individuals in 18–53 % of families.10–15 SADS deaths are often due to undiagnosed ion channelopathies, most commonly long QT syndrome (LQTS), Brugada syndrome (BrS) and catecholaminergic polymorphic ventricular tachycardia (CPVT). Family members of SADS victims may also be diagnosed with cardiomyopathy, including hypertrophic cardiomyopathy (HCM) and arrhythmogenic right ventricular cardiomyopathy (ARVC) where phenotypic expression in the sudden death case has been undetectable even at expert autopsy.12 If a post-mortem examination has not been carried out, the differential diagnosis of SUDS is wider, including a higher proportion of cardiomyopathy and additional familial diagnoses such as aortopathy and premature coronary artery disease due to familial hypercholesterolaemia. It must also be considered that some cases of SUDS may not be due to cardiac disease.
Specialist Cardiac Post-mortem
Familial evaluation begins with a comprehensive and expert post-mortem examination of the SUDS proband. It is recommended that all SUDS victims with an apparently normal post-mortem are evaluated by a specialist cardiac pathologist16 in order to detect subtle macroscopic and histological markers of cardiomyopathy that may not be diagnosed by a general pathologist, and to avoid over-interpretation of minor pathological findings that may lead to erroneous diagnoses of cardiomyopathy.
The need for expert input, working to internationally agreed guidelines and diagnostic criteria,16 is illustrated by the significant disagreement between diagnoses made by general and specialist cardiac pathologists. De Noronha et al.17 examined 200 consecutive cases of sudden cardiac death referred to a specialist pathology centre. The referring pathologist had made a provisional diagnosis in 158 cases and there was disagreement with the specialist cardiac pathologist in 41 % of these cases (kappa 0.48), with the general pathologist over-diagnosing cardiomyopathy, in particular ARVC, and under-diagnosing the structurally normal heart. In addition, Papadakis et al.18 demonstrated that familial evaluation in cases of SUDS with minor pathological findings at autopsy reveals a similar prevalence of channelopathy in first-degree relatives as in a contemporaneous SADS cohort. These findings include idiopathic left ventricular hypertrophy without myocardial disarray; fat within the free wall of the right ventricle without associated fibrosis to support ARVC;19 minor coronary artery disease without evidence of acute occlusion, ischaemia or prior infarction; and isolated lymphocytic inflammatory foci and minor ventricular dilatation. These features may be considered as common and should not automatically be considered as diagnostic of the cause of death. Therefore, such cases should be considered as SADS. It is also recommended that there is retention of fresh spleen or other suitable material for DNA extraction and post-mortem genetic analysis or ‘molecular autopsy’ in all SADS cases.1
Post-mortem non-invasive imaging has been shown to be useful in cases of non-cardiac death.20 When sudden cardiac death is due to ischaemic heart disease, computerised tomography (CT) coronary angiography can reliably identify coronary artery stenoses or occlusions,21 but is less useful in identifying ischaemic myocardium, the presumed trigger for sudden death. While cardiac magnetic resonance imaging (MRI) has a well-established role in the diagnosis of several cardiomyopathies during life, there are very few reports concerning post-mortem radiological imaging in non-ischaemic sudden cardiac death. At present, these methods have not been sufficiently validated to be considered routine, but may complement the traditional post-mortem in the future.
Molecular Autopsy
Since SADS is due primarily to undiagnosed channelopathies, post-mortem genetic analysis, ‘the molecular autopsy’, may identify a causative mutation in a responsible gene. This can then facilitate cascade screening in surviving family members, complement clinical evaluation and reassure those who are shown not to be carriers.1,22
The molecular autopsy has progressed since early reports.23,24 Studies analysing the main genes responsible for CPVT and LQTS have demonstrated diagnostic yields of up to 32 %.24–28 In the largest series to date, Tester et al. report the findings from molecular autopsy of 173 consecutive sudden cardiac deaths over a 12-year period.28 They used comprehensive sequencing of LQTS genes (KCNQ1, KCNH2, SCN5A, KCNE1, KCNE2) and focused sequencing of the CPVT1 gene (RYR2) and found causative mutations in 45 (26 %) cases. Positive findings were more common in those cases with exercise-related sudden deaths and in females.
Next generation sequencing may be of further benefit allowing a greater number of potentially causative genes to be tested. Bagnall et al. have reported whole exome sequencing-based molecular autopsy in 28 SUDS cases. In addition to three putative pathogenic rare variants in the more commonly analysed LQTS genes, six rare variants were found after including 25 further genes commonly associated with cardiomyopathies and ion channel disease (calcium channel subunits, CACNA1C and CACNA2D1; desmoplakin, DSP; sarcomere associated genes, MYBPC3 and TTN).29 There was, however, uncertainty over the pathogenicity of these variants, which were labelled as ‘variants of unknown significance’ (VUS). In addition, next generation sequencing coverage can be uneven and genetic yields will never reach 100 %.30 Therefore, a negative result does not exclude the presence of genetic disease or the need for familial evaluation where otherwise indicated. Thus the increased diagnostic yield and benefit of the next generation molecular autopsy will only be realised once there is improved interpretation of the VUS, and the clinical and genetic context of the family has been addressed.
Familial Assessment
Several studies have shown that family screening of SUDS and SADS relatives reveals evidence of inherited cardiac conditions in a significant proportion of family members screened10–15 with yields of 18–53 % reported. Tan et al. found that positive findings were more likely when a larger number of family members were investigated, and where there had been two or more sudden unexplained deaths below the age of 40 in a single family. There was a trend toward positive diagnoses seen more commonly when the proband died during exercise or with emotional stress, although this did not reach statistical significance.11
The guidelines advise the systematic evaluation of first-degree blood relatives with a focus on symptomatic relatives and obligate carriers. Assessment should ideally be carried out by, or discussed with, an appropriately experienced specialist. Initially the prior history, circumstances surrounding the sudden death and the family’s history should be evaluated31 as well as any ante-mortem electrocardiograms (ECGs). Each relative’s personal medical history should be assessed and a physical examination performed. A resting 12-lead ECG is recommended in all cases given its simplicity, wide availability and high diagnostic yield.11
Transthoracic echocardiography should be performed. This is principally to confirm a structurally normal heart, although a small minority of SADS relatives may be diagnosed with cardiomyopathy.12
Exercise Electrocardiogram
The exercise ECG is recommended as a first-line investigation.1 Increasing ventricular ectopy and/or tachyarrhythmia with exercise can be seen in various channelopathies or cardiomyopathies and is a diagnostic criterion in both ARVC19 and CPVT.1 However, two-thirds of patients with ectopy during exercise do not go on to have any other supporting evidence of cardiac disease.32 Findings of ST segment depression may support coronary artery disease, although the limitations of exercise testing for ischaemic heart disease are well-known and correlation with the overall clinical picture is required. The use of the exercise ECG to diagnose LQTS has been limited by difficulties in precise measurement of the T wave at rapid heart rates and by the significant artifact caused by exercise. More recently, Sy et al. found that QTc prolongation in the recovery phase after exercise can be used to accurately diagnose LQTS in suspected cases with a normal or borderline QT interval at rest.33,34 The results of this study were subsequently integrated into a modified Schwartz score, with a cut-off QTc ≥480 ms at the fourth minute of recovery.35 It has also been proposed that the exercise ECG may have a role in risk stratification in early repolarisation syndrome (ERS). The early repolarisation (ER) pattern is common in the general population and benign in the vast majority – although it has been associated with idiopathic ventricular fibrillation36 and an increased risk of sudden death.37 Bastiaenen et al. showed that in the majority of individuals with ER, J-point elevation was suppressed during exercise. ER with a horizontal ST segment where the J-point elevation persisted into exercise was associated with prior unexplained syncope.38 However, further investigation and validation is required before exercise testing is considered a risk stratification tool in ERS.
Investigation beyond the initial combination of resting ECG, echocardiography and exercise testing are class IIa and IIb recommendations.1 These include sodium channel blocker provocation testing, ambulatory ECG monitoring, signal-averaged ECG, epinephrine test and a fasting serum cholesterol level if no post-mortem has been carried out and premature coronary artery disease is suspected.
Brugada Syndrome and Ajmaline Provocation
The recent consensus document states that a diagnosis of BrS can now be made in “patients with ST-segment elevation with type 1 morphology ≥2mm in ≥1 lead among the right precordial leads V1, V2 positioned in the 2nd, 3rd or 4th intercostal space occurring either spontaneously or after provocative drug test with intravenous administration of class I anti-arrhythmic drugs.” The ECG changes seen in BrS correlate to abnormalities of the right ventricular outflow tract.39–41 Anatomically, this structure is commonly positioned superior to the standard V1 and V2 ECG electrode positions42 (right and left parasternal edges in the fourth intercostal spaces). The use of high right ventricular leads with electrodes in corresponding positions in the second and/or third intercostal space have been shown to increase sensitivity of observing a type 1 Brugada ECG pattern both in those with a spontaneous pattern and in the context of ajmaline provocation.43–45 Although, according to the previous BrS consensus statement, a type 1 ECG pattern was required in ≥2 leads,46 it has since been shown that the clinical profile and arrhythmic risk is similar in those with the type 1 Brugada pattern seen in a single lead.47 Savastano et al. have suggested that the new diagnostic criteria improve sensitivity while maintaining specificity.48
ECG changes in patients with BrS can be dynamic and therefore in those family members where initial investigations have been unremarkable, provocative testing with sodium channel blocking drugs is recommended (IIa). Ajmaline, flecainide, procainamide or pilsicanide can be used. Ajmaline has the benefit of a very short half-life minimising the length of the test and need for prolonged monitoring afterwards. It has a good safety profile, although rare cases of cholestatic hepatitis have been reported.49 The recommended dose is 1 mg/kg given intravenously over five minutes.46 Precipitation of a type 1 Brugada ECG pattern in one or more lead, in the standard or high intercostal space positions, constitutes a positive result.1 Due to the lack of a gold standard investigation to compare with and the lack of data regarding the effect of ajmaline in healthy controls, the specificity of ajmaline provocation in SADS family members is unknown and false positives cannot be excluded.
Epinephrine Infusion
Ackerman et al. first showed that epinephrine infusion (0.1 μg/kg/ minute) could be used as a diagnostic tool in LQTS with genotypepositive LQT1 patients demonstrating paradoxical QT prolongation.50 Shimizu et al. then showed that paradoxical QT prolongation differentiated genotype positive LQT1 patients with normal resting QTc intervals from normal controls.51 These early studies concluded that epinephrine infusion was both sensitive and specific for the diagnosis of LQT1, but caution must be exercised when extrapolating results from such well-defined patient groups. Krahn et al. reported results of epinephrine infusion given at 0.1 μg/kg/minutes in 170 consecutive cases comprising cardiac arrest survivors and relatives of sudden death victims, all with normal resting QTc measurements. An abnormal result, defined as a QT prolongation of ≥30 ms, was reported in 18 % of cases. Prolongation of 1–25 ms reported as a borderline result was seen in 14 % of cases. However, there was only modest correlation between an abnormal response to epinephrine and exercise, and only 20 % of individuals with an abnormal epinephrine response who subsequently underwent genotyping had a mutation identified. The authors therefore concluded that epinephrine testing is likely to be sensitive for LQTS but the specificity is questionable.
Epinephrine infusion can also be used for the diagnosis of CPVT. The presence of multifocal ventricular premature beats (VPBs) or bidirectional or polymorphic ventricular tachycardia (VT) is considered a positive result. Studies similar to those for LQTS have demonstrated variable sensitivity52–54 but abnormal results in controls are rare.51,55 However, the lack of a gold standard test or large systematic studies mean that, as for LQTS, the true specificity remains unclear.
Unexplained Cardiac Arrest
It is recommended that survivors of unexplained cardiac arrest (UCA) with documented ventricular fibrillation (VF) and their relatives are assessed in a similar manner to relatives of SUDS victims. Van der Werf et al. reported on the results of such assessment in 69 consecutive survivors of cardiac arrest.13 A definite or probable diagnosis was made in 42 survivors (61 %). The diagnosis was of an inherited cardiac condition in 31 survivors (45 %). This rate was understandably higher than in SUDS relatives in the same study – first-degree relatives have only a 50 % chance of carrying the same mutation; many of the conditions being sought show incomplete penetrance; and not all first-degree relatives come forward for assessment.
More recently, Kumar et al. found a discrepancy between the yield of investigations in SADS relatives (18 %) compared with UCA relatives (62 %) despite similar protocol of evaluation.15 Some methodological factors contributed. For example, sodium channel provocation was only undertaken in a small proportion of relatives and high ECG leads were not used. Cardiac arrest survivors were excluded if coronary disease, valvular disease or impaired LV function were identified, yet the proportion of probands with other structural heart diseases such as HCM or ARVC was not specified. These would have been excluded in the SADS cases by definition. ERS was also diagnosed in 6 % of families. While the latest recommendations state that ERS can be diagnosed in a SCD victim where an ante-mortem ECG has shown the ER pattern,1 many SADS probands never have an ECG since the majority are asymptomatic until their fatal episode.31 The high prevalence of ER in the general population,37 especially in young adults,56 and the lack of available risk stratification tools also contribute to making the diagnosis of ERS challenging and controversial in asymptomatic SADS relatives.
Management
The majority of individuals assessed will have reassuring findings. If fully grown adults, patients can be discharged without further follow-up. Children and adolescents may need periodic re-evaluation due to age-related penetrance of many of the conditions in question.57 Those with borderline or inconclusive initial investigations may also benefit from repeat assessment, although it is important to be clear that no formal diagnosis has been made.
The management of individuals with positive diagnoses should follow recognised guidelines for the specific conditions,1 which will frequently involve lifestyle modification and serial monitoring. Initial reports suggested a high rate of intervention with beta-blockers in 51 % and implantable cardioverter defibrillator (ICD) in 11 % of affected individuals.12 More recently, Caldwell et al. reported that beta-blockers were initiated in 40 % of affected relatives and 9 % received a primary prevention ICD.58
Psychological Support
The sudden death of a young family member can have multiple psychological effects on a family and it should be noted that familial assessment is often carried out at a time when grieving may still be prominent. In addition, diagnosis of a heritable condition can engender feelings of guilt, anxiety and depression, particularly in the parents of a sudden death victim. Access to psychological support should be available for families and good relationships with patient support groups and charities working in the field of young sudden death can be beneficial.
Summary and Conclusion
Familial assessment of SADS family members is an important tool in reducing the burden of sudden death in the young. A significant proportion of family members will be diagnosed with cardiac genetic disease, the majority of whom can be offered effective treatment and lifestyle modifications without the need for ICD implantation. Negative findings offer reassurance to unaffected family members. Simple non-invasive tests remain the cornerstone of investigation with questions over specificity in SADS family members existing over many of the more recently described protocols. The molecular autopsy may offer a complementary role in the evaluation of families.