







Abnormalities in electrical rhythm were studied by Einthoven at the start of the 20th century. In the 1940s, studies by Bozler et al.1 described contractile signals that appeared to be ‘triggered’ heart beats. Today we use the term delayed afterdepolarisations (DADs) to refer to oscillations in voltage that follow a driven action potential.
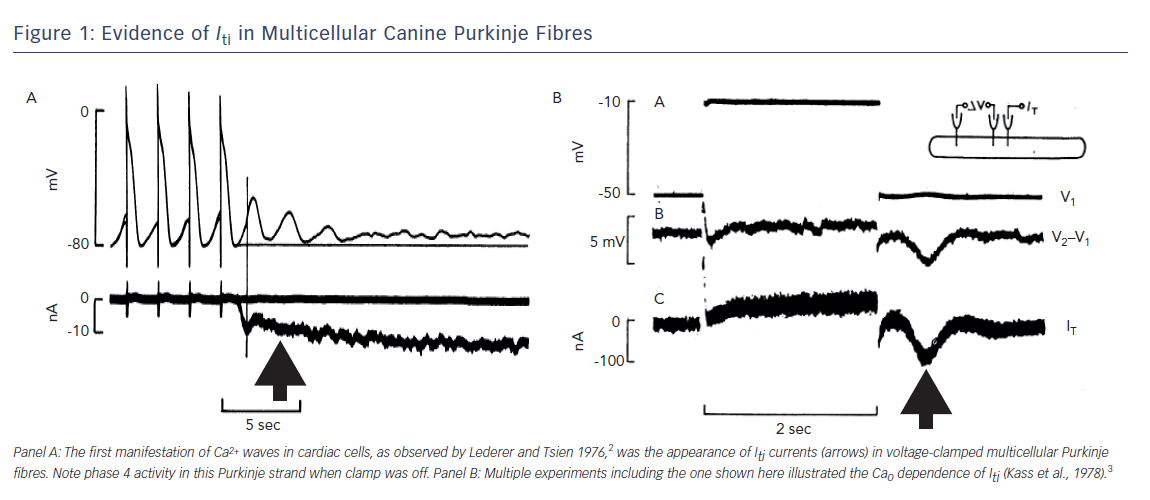
In the mid-1970s, progress was made when Lederer and Tsien developed a method to study the underlying electrical mechanism of DADs2 (see Figure 1). In a voltage clamped, multicellular canine Purkinje fibre, the transient depolarisation of the resting potential of the fibre was found to be due to a transient inward current (Iti) (see Figure 1A). Many initially challenged this idea but these authors went on to show that Iti was not an artifact and that the Iti they recorded in Purkinje fibres was Ca2+ dependent (see Figure 1B).2,3 This was a relatively new concept for cardiac electrophysiology; that is, the idea that Ca2+ inside the cell could feed back and affect the electrics of the cell’s membrane. In a recent review this was referred to as reverse mode excitation–contraction (EC) coupling.4
Here we will discuss the Ca2+ wave and address the question: ‘Is it like the electrical wave with which we are all familiar?’
Functional Anatomy
A propagating electrical wave utilises the energy of the chemical gradients set up by the cardiac sarcolemma.5 Electrical waves rely on activation of a series of ion channels (eg. Na channel proteins) for forward propagation of the wave.
Propagation of a Ca2+ wave also depends on the energy stored in the myocyte. But in this case the energy comes from the presence of Ca2+ stored in the sarcoplasmic reticulum (SR). The SR is a specialised intracellular membrane structure that in a myocyte stores Ca2+ that has been pumped into it by a SR membrane pump, SERCA2. In the absence of Ca2+ influx through the plasma membrane or mischievous Ca2+ wandering the cytosol, the Ca2+ in SR stays in the SR. This is because the SR ligand-operated Ca2+ channel, the ryanodine receptor channel (RyR), which guards this SR Ca2+ store, has a low probability of opening.
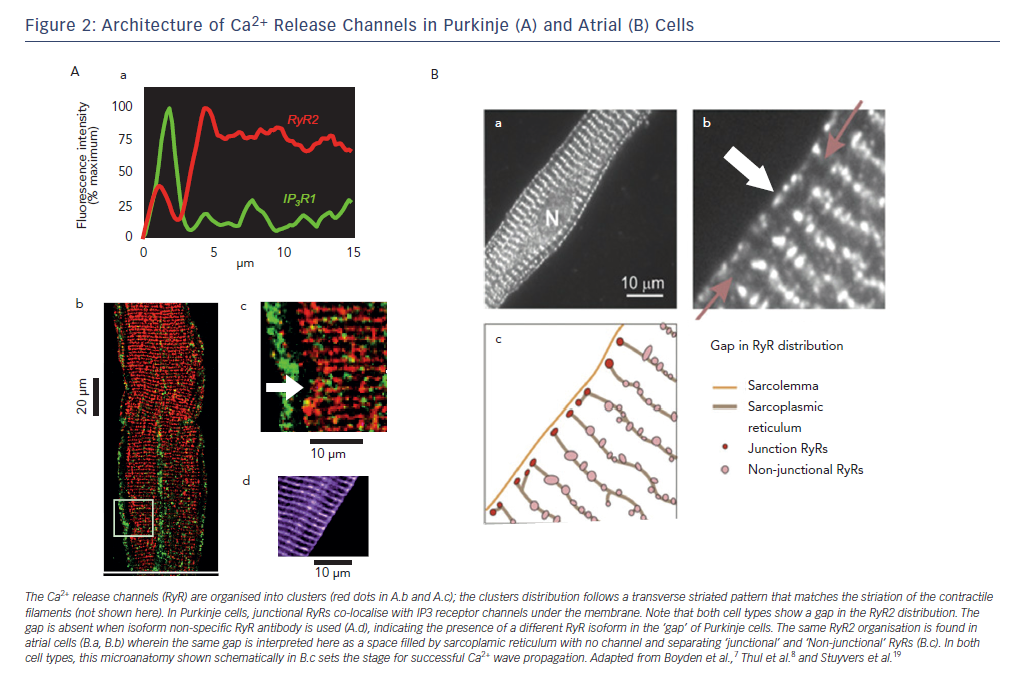
Interestingly, just as surface membrane ion channels (eg. Na channels) are positioned in a specific array6 to provide for smooth electrical wave propagation, RyR channel proteins in myocytes, Purkinje and atrial cells are clustered and aligned in a specific micro-anatomic pattern (see Figure 2).7,8,19 Presumably, and particularly in the tubulated structures of ventricular myocytes, this specific patterning is to allow for uniform Ca2+ release from SR during the action potential (forward mode EC coupling). The orderly pattern of RyRs on the SR sets up a series of potential release sites of Ca2+ in the cell.
Ca2+-induced Ca2+ Release
Fabiato’s work on the properties of the cardiac SR provided a potential explanation for spontaneous Ca2+ release in mechanically skinned cells in which the SR and RyR were intact and excessive Ca2+ loading of the SR caused spontaneous Ca2+ release.9,10,11 The mechanism for increased probability of opening of RyR when the SR is heavily loaded with Ca2+ is still uncertain, but suggests that the RyR channel is sensitive to both cytosolic and luminal [Ca2+] of the SR. Hence, the oscillatory character of a triggered arrhythmia in myocardium with a high cellular Ca2+ load may be due to further increase of Ca2+ entry into the cells during driven action potentials, which causes even more Ca2+ loading of the SR. So as soon as the release process has recovered after the electrically evoked Ca2+ release, the overloaded SR again releases a fraction of its Ca2+ into the cytosol. The requirement that the Ca2+ release mechanism must recover first (refractoriness) would explain the presence of a delay between aftercontractions and afterdepolarisations and the preceding beat.
Ca2+ waves occurring in cardiac cells depend on the regenerative production of a diffusible molecule that triggers Ca2+ release from adjacent SR stores. Cytosolic Ca2+ is one such ion and thus the process is called Ca2+ induced (intracellular) Ca2+ release (CICR) (see Figure 3B).12 This schematic shows Ca2+ wave propagation from one RyR cluster to another. Calsequestrin (CASQ), a Ca2+ binding protein, is found in the SR lumen and aids wave propagation inside cells (see Figure 3C). The released Ca2+ constitutes a leak from the SR and tends to reduce the overload. This phenomenon has been observed in different forms, all of which fall under the general definition of Ca2+ leak: increased probability of opening of RyR in lipid bilayer experiments,13 a biochemically detectable loss of Ca2+ from the SR;14 Ca2+ sparks in isolated cells and muscle;15,16 micro Ca2+ waves in isolated cells and muscle13,14 and Purkinje cells after infarction;7,19 and multicellular cellular Ca2+ waves.17–19 The threshold for Ca2+ leak is reduced in some arrhythmogenic mutations of the RyR,13 CASQ20 and in acquired dysfunction of the RyR such as in congestive heart failure and post MI.7,21–23
Intracellular Ca2+ waves can be seen in normal canine atrial and Purkinje myocytes during forward mode EC coupling (see Figure 4).7 Here, a line of Ca2+ release is seen peripherally just after the plateau of the AP and this Ca2+ then via CICR, sets up a Ca2+ transient that moves to the core of the Purkinje cell (see Figure 4).
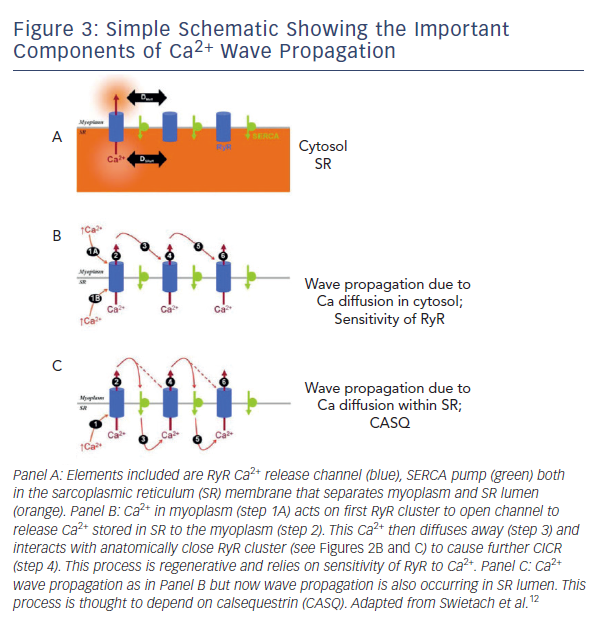
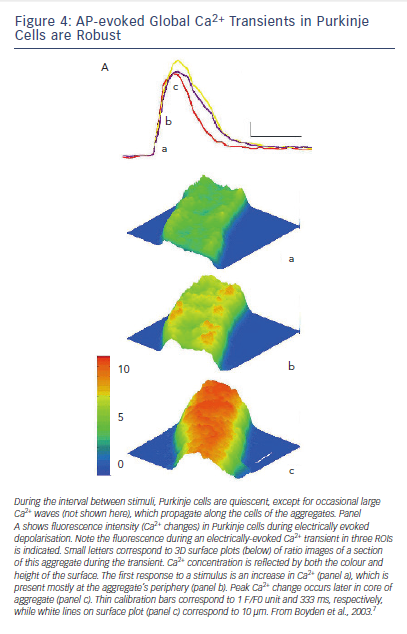
Figure 5A shows that spontaneous Ca2+ waves occur often during diastolic intervals in Purkinje cells dispersed from the infarcted heart.7
In some cases the waves formed varied oscillatory changes in voltage as
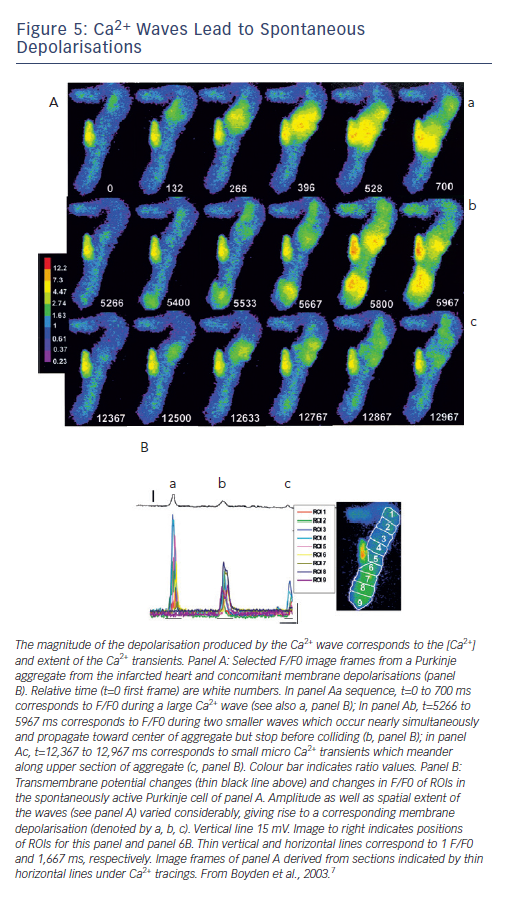
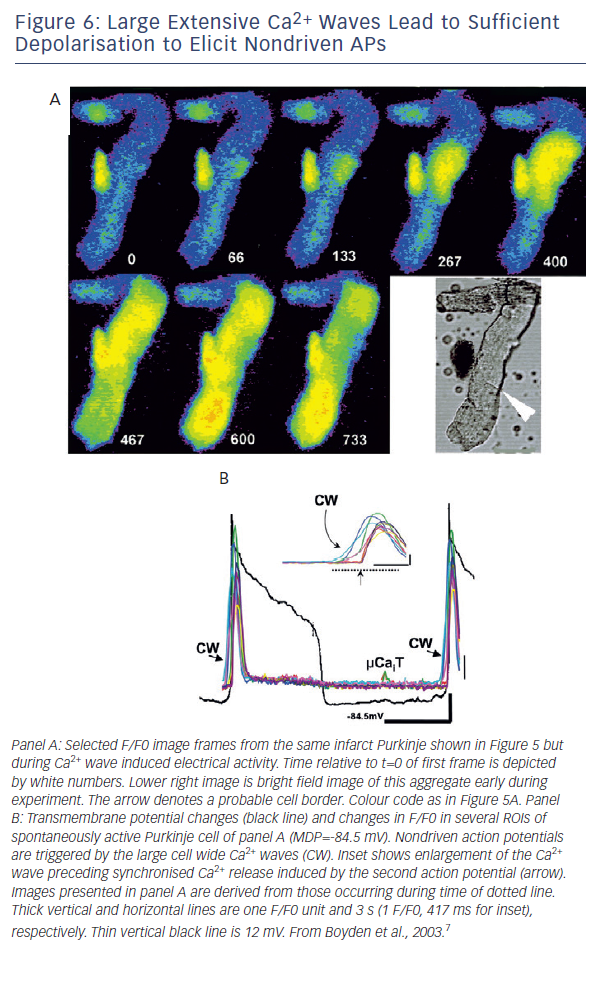
Ca2+ of wave is pumped out of cell via the sodium-calcium exchanger (Iti) (see Figure 5B). However, in the same cell the oscillatory voltage change is large enough to reach threshold, triggering a nondriven AP (see Figure 6). In these cells the small voltage signals and triggered activity are sensitive to an agent that blocks Ca2+ release of the RyR protein, ryanodine.
Propagation Between Cardiac Cells
Intercellular electrical transmission occurs via a set of ion channel proteins and specialised membrane structures called gap junctions.24 Each channel is formed by close apposition of two hemichannels each of which is in an opposing cell.25 Gap junctions can provide passage of many molecules (cAMP, Ca2+, IP3, ATP).26–28 In cardiac cells gap junctional conductance can be regulated acutely by pH, Ca2+, cAMP and cGMP.29 Therefore Ca2+ ions can flow through gap junctions as well as inhibiting gap junctional conductance.
Since Ca2+ waves propagate along the cell, it is important to know whether they propagate between cells via gap junctions. Many have observed Ca2+ waves passing between two cardiac cells30 and have assumed a role for gap junctions. Ca2+ ions released upon RyR activation can travel as a wave across cells31 and propagate to adjoining cells via gap junctions.19 In cells transfected with both
connexin 43 (Cx43) and RyR receptors the propagation of Ca2+ waves between cells was sensitive to octanol.32 Furthermore, in this experimental cell model, both Ca2+ wave propagation and gap junctional conductance between paired cardiac cells are related to the state of tyrosine phosphorylation of Cx43.33 How the Ca2+ wave crosses
the gap junction is unknown but extracellular disulphide bonds of
the Cx43 proteins between the adjoining cells appear critical for
wave propagation.32
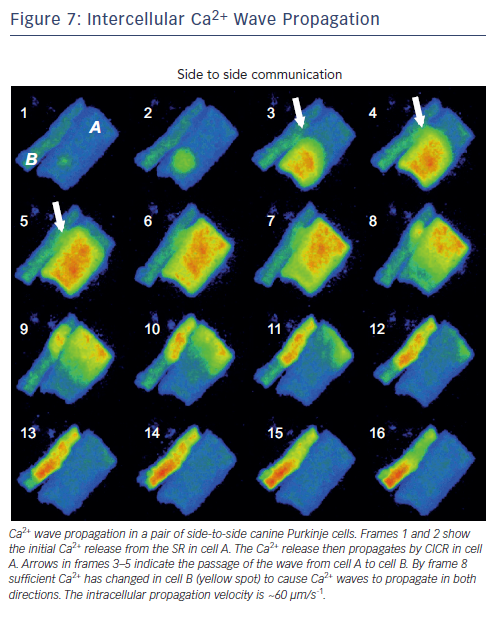
Arguably the occurrence of Ca2+ wave propagation from one cell to another is not a frequent event, but when one does happen, it appears to be due to a CICR mechanism (see Figure 7). In adult rat cells, Li et al.30 assert that Ca2+ wave propagation between cells mostly occurs at side-to-side junctions and the ultrastructure of the connections between the gap and SR release units is critical. Propagation failure occurs when distance between the disc membrane and neighbouring SR release unit is too large, such as occurs at end-to-end junctions.
At the tissue level, the subcellular Ca2+ dynamics combine with cell coupling and tissue architecture to generate multicellular Ca2+ wave dynamics. We understand focal electrical excitations due to triggered activity but are the dynamics of Ca waves similar? Spontaneous Ca2+ releases that triggered Ca2+ waves have been mapped in both normal and failing heart tissues34 (see Figure 8).35 Notably each
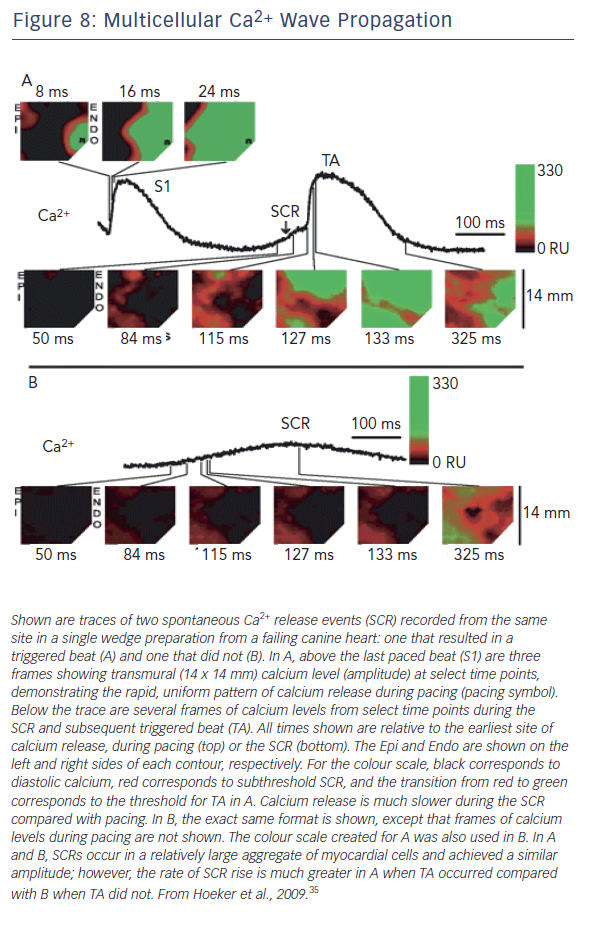
spontaneous event occurred in a region of myocardium comprised of many cells (~3000 cells).34 In failing myocardium it is the rate of rise of the Ca2+ releases (waves) rather than their amplitude that is associated with triggered beats. In atria from CASQ-/- mice, the runs of APs during nondriven electrical activity were always preceded by rises in Ca2+, however total time of atrial activation increases with number of beats.36
Refractoriness
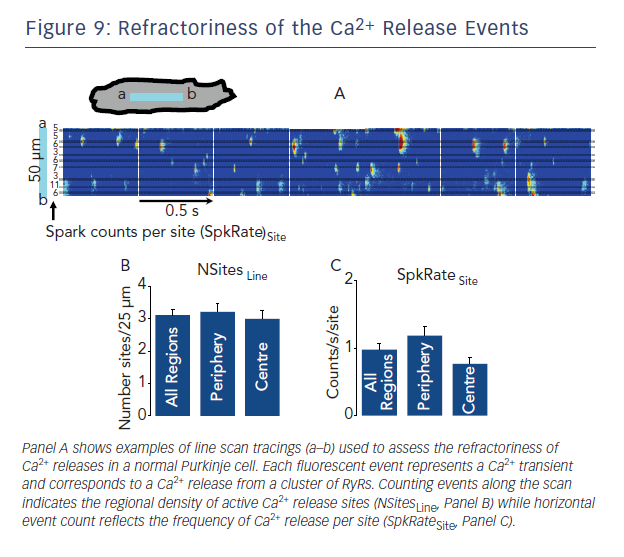
Electrical refractoriness is clearly related to time course of action potential repolarisation as well as the status of the sodium channel. In forward mode EC coupling, after the cellular action potential evoked Ca2+ transient due SR Ca2+ release, time is needed before a second Ca2+ release occurs of similar amplitude. Thus there is also a recovery process of Ca2+ release in cardiac cells that is independent of membrane voltage. In the electrically stimulated cell, the recovery of the SR Ca2+ release process or refractoriness is determined by recovery of the L-type calcium channel influx as well as the time course of the SR refilling. The latter can be examined by assessing the interval between spontaneous Ca2+ sparks occurring at the same release site (see Figure 9). Ca2+ spark termination is due to local depletion of Ca2+ within junctional SR. The time between one spark and another is related to the ryanodine sensitivity or threshold for Ca2+ release. How fast junctional SR refills after depletion is important for the recovery of both spark amplitude and CICR or Ca2+ wave formation.37 Shortened refractoriness of this process has been seen in both acquired (post MI)38 and genetic disease.39 For example, loss of calsequestrin (CASQ) in SR of cells in some CVPT patients produces fast SR refilling and greater likelihood of a trigger for re-release40 and Ca2+ waves.
Conclusion
While cellular electrical events and Ca2+ waves can occur independently of each other, it is when they interact and feed back on each other that complicated arrhythmogenic behaviour can occur (eg. alternans).4 Each physiological process has its own mechanisms of initiation, propagation and refractoriness and thus would be expected to have its own possible targets for effective therapeutic agents. For example, CamKII,41 sodium-calcium exchanger protein42 and EHD343 proteins have all emerged as possible targets for Ca2+-dependent arrhythmias. n