










Anisotropy is the property of directional dependence. In cardiac tissue, conduction velocity (CV) is anisotropic, that is, the magnitude of CV depends on the direction of a wave of activation. Anisotropic conduction facilitates prompt and synchronised cardiac chamber activation. In disease states, anisotropic conduction is implicated in the genesis of pathological arrhythmias. It can determine unidirectional conduction block (UCB), which is a prerequisite for re-entry, the likelihood of propagation of ectopic foci of activation and the motion and stability of re-entrant activation patterns. The response of cardiac tissue to treatment, such as pacing, and the ability of a high-voltage stimuli to terminate fibrillation through the generation of virtual electrodes are dependent on the anisotropic properties of the myocardium.1,2 Therefore, anisotropic conduction is an important property of cardiac tissue, disease and response to treatment of arrhythmias. In the present study, we discuss the mechanisms determining anisotropic conduction within cardiac tissue, the contribution of anisotropic conduction to the mechanisms underlying pathological re-entrant arrhythmias and the reported options for assessing anisotropy.
Factors Responsible for Anisotropic Conduction
CV varies in different locations within the heart. The magnitude of CV within cardiac tissue demonstrates directional dependence, that is, anisotropy, with the maximum speed of conduction being observed parallel to the direction of myocytes (longitudinal CV) and the slowest speed of conduction perpendicular to the myocytes (transverse CV).3,4 A further layer of complexity arises when conductivity is considered in three dimensions. At a cellular level, myocytes are transversally isotropic, that is, CV is equal in any direction in the plane perpendicular to the principal axis of the myocyte; however, this does not hold true when conduction is considered at the tissue scale, where the laminar structure of the tissue results in orthotropic conduction, that is, variation in conductivity in each of the three dimensions.5,6 Much of the literature pertaining to anisotropic conduction makes the assumption, often implicitly, of transverse isotropy of conduction, which must be considered a limitation in light of these data regarding orthotropy. However, having acknowledged this limitation, the consideration of anisotropy in two dimensions yields important data. The ratio between the maximum and minimum CV observed is known as the ‘anisotropy ratio’ (ARCV) and indicates the magnitude of CV anisotropy within that tissue. Other characteristics of cardiac tissue also demonstrate anisotropy, including the conductivity of tissue (a parameter that is frequently considered in cardiac modelling studies) and the physical structure of the tissue.7,8
Several factors have been identified that contribute to CV anisotropy. These include cell size, cell shape and gap junction distribution. Various experimental setups have been established to attempt to separately measure longitudinal and transverse CV.9 One of the challenges in doing so has been to separate the impact of intrinsic tissue anisotropy from the impact of wavefront curvature, which has a profound effect on CV and is influenced by anisotropy.10 The impact of wavefront curvature presents significant challenges for the assessment of ARCV in vivo.9 Along with the complex 3D alignment of myocytes within the human heart, these challenges have prevented routine assessment of ARCV within intact cardiac chambers.11
Cell Shape and Size
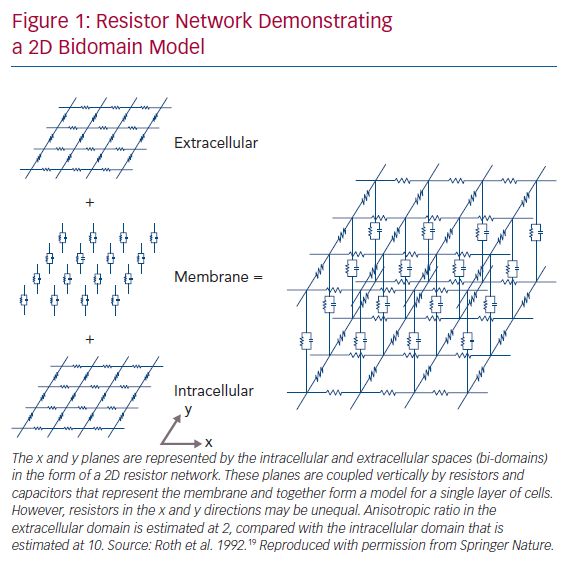
Cardiac myocytes are elongated cells with a cylindrical shape. They are oriented in bundles of parallel cells that then form into laminar sheets, rendering the microstructural architecture of human cardiac tissue anisotropic.12 Early experimental observations on the anisotropic nature of cardiac conduction noted myocyte orientation as the principal determinant of the direction of the maximal CV observed.13 In addition to experimentally demonstrating anisotropic conduction of the myocardium, Clerc also highlighted the presence of unequal resistivity anisotropy ratios in the intra- and extra-cellular domains, which was confirmed in subsequent studies.13,14 This key observation prompted further experimental and theoretical work in the coming years with significant implications for understanding complex propagation and polarisation patterns in 2D and 3D cardiac tissue, and was instrumental in the development of the bidomain model of cardiac electrophysiology (Figure 1).15–19
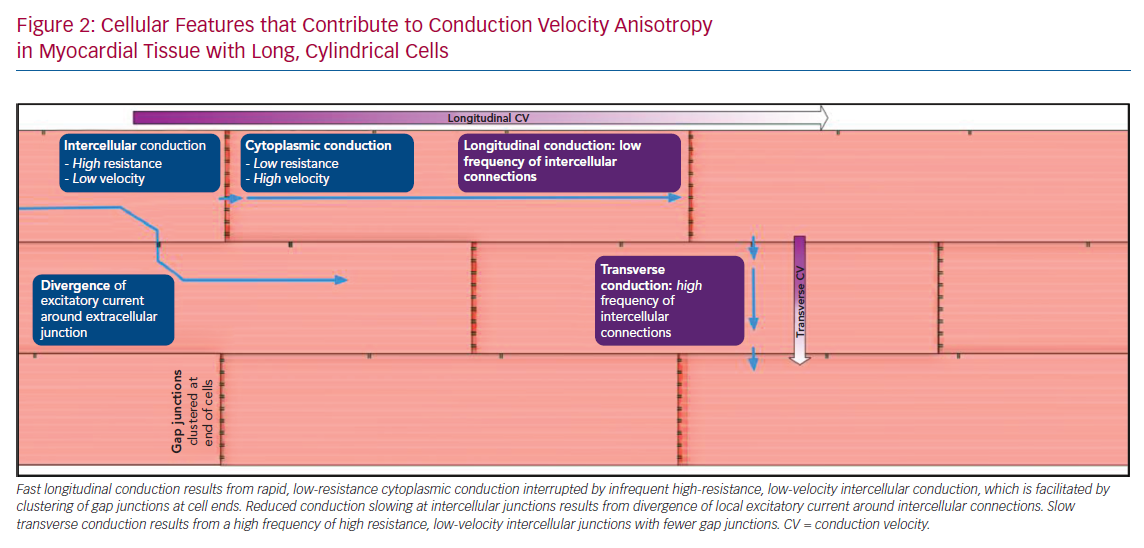
As demonstrated in Figure 2, a wave of conduction travelling perpendicular to the principal axis of a myocyte (transverse direction) will meet a greater number of cellular interfaces per unit length than will be encountered in the direction parallel to myocyte direction (longitudinal direction). Cytoplasmic conduction along the length of the myocyte is rapid with low resistance, whereas low-velocity, high-resistance conduction occurs across the gap junctions that electrically couple adjacent cells. This distinction is an important determinant of CV anisotropy. When isolated single chains of myocytes are considered, conduction slowing at cellular junctions results in discontinuous or saltatory conduction, and >50% of conduction time may occur at the small distance across gap junctions, with the remainder of conduction time being due to cytoplasmic conduction.20,21 However, Figure 2 further demonstrates the reduction of this effect in 2D laminar tissue due to lateral cell connections that permit divergence of the local excitatory current around the junction, effectively speeding up conduction across the cellular interface.21 Considering propagation in the longitudinal direction, it is also evident that a depolarising wave travelling in a longer cell will encounter a lower frequency of cellular interfaces than a wave travelling in a shorter cell, with a consequent increase in longitudinal CV in longer cells, and therefore, anisotropy. Experimental and modelling studies indicate that pathological changes in cell shape and size affecting the length–width ratio will have a consequent impact on anisotropy of conduction.22 Computational simulation of the activation of representative blocks of tissue confirms maximum propagation speed along the axis of myocytes, but also reveals additional levels of complexity to propagation patterns in 3D.5,6
Myocardial Fibrosis
Myocardial fibrosis is a common consequence of many human cardiac pathologies, and may be classified as reactive fibrosis, which is the result of increased collagen deposition, or replacement fibrosis (‘scar’), in which collagen replaces injured myocytes.23,24 Collagen deposition may be in the form of discrete regions of dense collagen without any viable myocytes (‘compact’ fibrosis), an increase in the extracellular matrix (‘interstitial’ fibrosis) or due to an intermingling of myocytes and stretches of collagen fibres.24 Different patterns of fibrosis appear to affect conduction and conduction anisotropy differently. Interstitial fibrosis, predominantly separating the longitudinal cellular bundles, results in the most pronounced decrease in transverse CV in the human heart, and an increase in ARCV.25 In this situation, enhanced microstructural discontinuities (observed in an animal model under experimental conditions) result in dissociated conduction with longer conduction times and a zigzag pattern, as well as promoting non-uniform action potential properties.26–28 These characteristics of transverse conduction are due to the tortuous route that a propagating wave must traverse, in addition to electrical uncoupling in the transverse direction, due to increased interstitial resistance to current flow.29 These changes amplify the ARCV of tissue, which favours the initiation of re-entry and will be discussed later.4
Gap Junctions
Gap junctions electrically couple adjacent cells by behaving as selectively permeable ion channels. The conductivity of gap junctions (a factor of the density of gap junctions and their individual behaviour) is a major determinant of intercellular resistivity. This in part determines CV, affecting the extent of delay encountered at the intercellular junction. Gap junctions cluster at the longitudinal end of myocytes, but are also found along the length of cells, electrically coupling cells in the transverse direction.30 Pathologically, the redistribution of gap junctions can result in a decrease in density of gap junctions at the longitudinal cell ends, and an increase along the shaft of the cells, known as ‘lateralisation’.30 Furthermore, an overall reduction in the expression of gap junctions can be seen in ventricular tissue. These findings have been observed in a range of pathological processes associated with arrhythmia, including dilated cardiomyopathy and ischaemic cardiomyopathy.31,32 When the gap junction protein Cx43 was reduced by 95% in a murine experimental model, ventricular tissue demonstrated decreased CV and increased ARCV, which resulted in greater susceptibility to ventricular arrhythmias.33 Full-thickness gap junction distribution disturbance was seen in an early post-infarct canine myocardium in which ventricular tachycardia (VT) was inducible and co-localised with the central common isthmus of these VT circuits, while only partial-thickness distribution abnormalities were observed in non-inducible hearts. In atrial tissue, genetically determined reduction in Cx40 and Cx43 function has been identified in patients with AF in the absence of any predisposing conditions. Furthermore, experimentally promoting Cx43 expression in a porcine model of AF restored CV, with a resultant decrease in susceptibility to AF.34–37 While these observations suggest that pathological changes in gap junction behaviour would be an important determinant of changes in CV and CV anisotropy in disease, other experimental evidence suggests that it has a relatively modest impact on changes in CV anisotropy.22 This may reflect an excess of gap junctions, providing a buffer that minimises the impact of redistribution and differences in function (conductivity) of the redistributed gap junctions in the pathological state. The surrogates used to identify the distribution of gap junctions (usually done through immunohistochemical analysis of the localisation of Connexin proteins) may also not accurately identify the location of the functional gap junction components.30,38 Resolving the discrepancy between the observation of remodelled gap junctions in clinical conditions associated with arrhythmia against experimental evidence suggesting a modest impact on CV with the observed changes in gap junction distribution has remained challenging. A further possible explanation may be the potentiation of changes in gap junction effects by increases in interstitial volume. In an experimental model of increased interstitial volume (which is observed in a broad range of cardiovascular pathologies), gap junction blockade resulted in slowed conduction and increased ARCV, which was associated with an increased susceptibility to arrhythmia that was not seen in controls.39,40 Outstanding issues remain surrounding the magnitude of the effect in gap junction remodelling and other conditions required to unmask the arrhythmogenic effects. However, current data suggest that the consequence of changes in gap junction function in disease predispose to arrhythmia in a range of conditions.
Functional Determinants of Anisotropy
In addition to structural determinants of CV and ARCV, there are important functional contributors to these characteristics. The high density of sodium channels found at the intercalated discs between myocytes represents an important modifier of conduction at the intercalated discs, and thus, conduction along the axis of the myocyte.41,42 Activation of the sympathetic nervous system increases longitudinal CV, but not transverse CV, therefore increasing ARCV in a porcine ventricular myocardium. This effect was shown to be abolished by gap junction blockade, demonstrating the importance of the functional modulation of anisotropic conduction.43
The clinical significance of alterations in sodium channel function is demonstrated through the loss of function mutations of the sodium channel protein type 5 subunit alpha (SCN5A) gene, resulting in reduced CV, as is seen in Brugada and Lev–Lenègre syndromes. Prior in vivo studies have demonstrated that reduced CV in the context of normal heterogeneities within the right ventricle predisposes to vulnerabilities of re-entrant tachycardias that may precipitate ventricular fibrillation, and consequently, sudden cardiac death.44
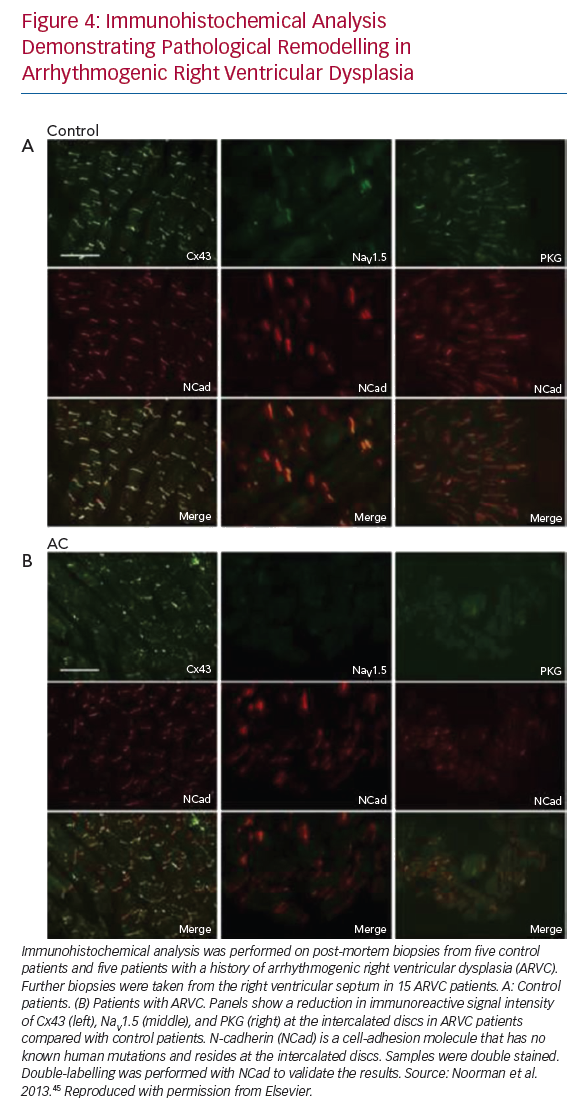
More recently, a reduction in the number of cardiac sodium channels was shown using immunocytochemistry of cardiac tissue from patients with a diagnosis of arrhythmogenic right ventricular cardiomyopathy (ARVC).45 The same study also demonstrated reduced signal of the gap junction protein Cx43, previously discussed as an important determinant of changes in CV and CV anisotropy in disease. These observations likely contribute to the increase in arrhythmia vulnerability seen in ARVC.
Variations in Anisotropic Ratio and Conduction Velocity in the Heart
Atrium
Specific regions of preferential conduction within the atria appear to be optimised for conducting a wave of depolarisation to facilitate an orderly sequence of activation in the atria. These regions include the crista terminalis, running from the sinus node to the eustachian valve and giving off trabeculations that facilitate right atrial activation and left atrial activation via Bachmann’s bundle.46 These regions demonstrate both faster CV and increased ARCV (in some cases transverse conduction is absent) than in surrounding atrial tissue.47,48 ARCV of up to 10 has been recorded in these regions.49 In the crista terminalis, pronounced anisotropy fulfils the functional requirement of permitting appropriate timing of atrial activation. ARCV in atrial body tissue has been less commonly reported, but in preclinical and clinical experiments where it has been quantified, it is lower than that recorded in either the crista terminalis or Bachmann’s bundle.50,51 Myocytes in the heart demonstrate patterns of organisation that may be seen when tissue is examined macroscopically.
Ventricular Tissue
The highest speeds of conduction found within the heart are in His-Purkinje tissue.52 These insulated cells are optimised for rapid longitudinal conduction of a depolarising wave exiting the atrioventricular node to a large mass of ventricular tissue.53 The rapid dispersion of a wave of activation permits synchronized ventricular contraction, obviating the need for rapid conduction between ventricular myocytes, and therefore, overcoming the relatively slow conduction between adjacent ventricular myocytes. The insulation of these tracts by a fibrous sheath prevents dispersion of current in a transverse direction (beyond the strand enclosed in a fibrous sheath).54 In contrast, CV in ventricular tissue is among the slowest within the heart and demonstrates a correspondingly low anisotropy ratio (ARCV). Under experimental conditions, mammalian ventricular tissue has a maximum longitudinal CV around 0.5–0.6 ms–1, whereas transverse CV is estimated between 0.15 and 0.2 ms–1, and thus the anisotropy ratio is between 2.5 and 4.13,55 In ventricular tissue, myocyte orientation seems to be optimised for mechanical efficiency, rather than for rapid dispersion of a wave of activation, which is instead facilitated by the cardiac conduction system tissue that rapidly disperses the wave of depolarisation, resulting in synchronised ventricular contraction.56
Anisotropic Conduction as a Substrate for Arrhythmia
Re-entry is a key mechanism underlying the majority of clinically important arrhythmias responsible for affecting patient prognosis or symptom burden. This may take the form of a stable and mappable re-entrant circuit, such as those seen in post-infarct VT, or may be a complex interaction of one or more unmappable re-entrant circuits, such as those seen in AF, the exact nature of which remains to be definitively established. Despite ongoing uncertainty surrounding the activation patterns in AF, it is acknowledged that re-entrant activation of the atria plays a key role.
Variations in Anisotropic Ratio and Conduction Velocity in the Pathological Heart
Examples of enhanced ARCV in pathological conditions have been reported, including during right atrial assessment in patients with chronically stretched atria secondary to mitral stenosis, who demonstrated increased ARCV at the crista terminalis compared to controls. This represents an example of pathological substrate remodelling in patients with a condition associated with a high incidence of AF.57 A similar increase in CV anisotropy has been observed in atrial tissue from patients undergoing surgical AF ablation.58 Optical mapping has demonstrated heterogeneously decreased CV and increased ARCV within left ventricular tissue from end-stage heart failure patients, as well as the increased incidence of transverse conduction block, reflecting substrate changes that may contribute to the increased incidence of re-entrant arrhythmia in this group.59 High-resolution activation mapping of post-infarct VT circuits in a porcine model suggest that extreme slowing of conduction perpendicular to the isthmus is present, with relatively normal CV within the isthmus, in a pronounced example of anisotropic conduction during a sustained arrhythmia.60
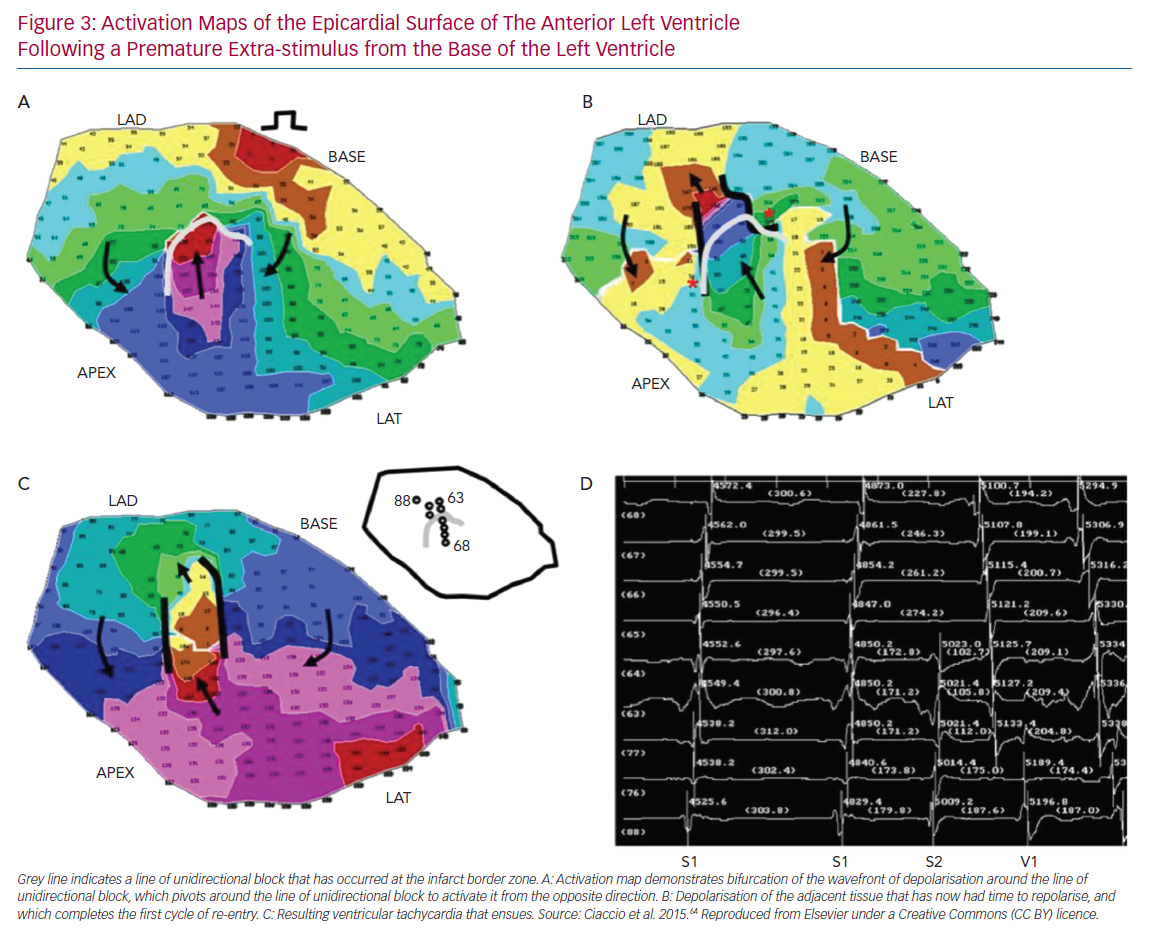
Unidirectional Conduction Block
UCB is a prerequisite for the development of re-entry.61 A variety of mechanisms may give rise to UCB, including functional asymmetry of the cardiac action potential, local heterogeneity of tissue excitability and discontinuities in tissue structure. Simulation studies demonstrate that local excitation within a homogeneous excitable media may interact with the tail of a preceding wave of depolarisation and result in UCB. In order to do so, the local excitation must be critically timed to fall within a vulnerable window, which may be defined in terms of membrane voltage, time or space.62 While UCB may occur in homogeneous media due to the intrinsic asymmetry of the action potential (termed ‘functional heterogeneity’), the width of the vulnerable window may be significantly expanded by heterogeneity of the electrical properties of tissue, including the availability of Na+ channels (excitability), cell-to-cell coupling (connectivity) and the behaviour of K+ channels (repolarisation).63 Furthermore, structurally determined source–sink mismatch at the transition from a small to a large mass of excitable tissue may also give rise to UCB, as propagation fails in the direction of a rapidly expanding volume of excitable tissue.64 The anisotropic behaviour of tissue has been demonstrated to promote UCB. In post-infarct VT, re-entrant circuits may be established by the appearance of an arc of conduction block, with subsequent activation of tissue distal to the initial arc sufficiently late that it has recovered excitability and depolarisation can occur (Figure 3).65,66 If an arc of conduction block occurs in the longitudinal direction relative to myocyte orientation, that is, in the direction of maximal CV, slower conduction occurs in the transverse direction (a function of the intrinsic difference between transverse CV and longitudinal CV due to tissue anisotropy, as well as wavefront curvature introduced by the arc of block). This allows additional time for recovery of excitability of tissue distal to the arc, such that re-entry is more likely to be established than if the initial arc of conduction block was in the transverse direction.55 Similar re-entrant patterns have been demonstrated in atrial tissue, identifying anisotropy of conduction as a key mechanism promoting re-entry.67 Recently, Anter et al. used high-resolution mapping of re-entrant VT circuits to show that a key substrate in the sustenance of the arrhythmia was conduction slowing at the entrance/exit of the VT isthmus.60 An important component of this was the highly curved nature of the wavefront propagation in these regions, transitioning from parallel to the faster fibre orientation (along the isthmus direction) to propagate transverse to it in order to loop back around and re-enter.60
Robustness of Conduction
The effect of pathological remodelling on the robustness of conduction, quantified as ‘safety factor’, and indicating under what circumstances conduction will fail, remains uncertain, although alterations in mechanical and electrical coupling of cells at the intercalated discs have been postulated as potential mechanisms. Figure 4 demonstrates disturbance in the immunoreactive signals of connexin 43 and sodium channels following pathological remodelling of the right ventricle in patients ARVC. These changes have previously been found to be associated with reduced CV, and have an increased susceptibility to the initiation and perpetuation of arrhythmias.45 Theoretical studies and experimental data suggest that uncoupling cells in the transverse direction may result in extremely slow yet robust conduction, an effect which would promote the development of re-entry.49,68 Other investigators have demonstrated more robust conduction in the longitudinal direction under experimental conditions as a result of cellular uncoupling.69 In fibrotic human left ventricular tissue, transverse conduction block was seen more frequently than longitudinal block.59 Differences in methodology may explain some of the discrepancies in the results, and it is possible that different mechanisms of remodelling have different effects on transverse conduction safety. At present, the existence of a consistent effect of pathological remodelling on the differences in robustness of conduction in the longitudinal versus the transverse direction remains uncertain.
Anisotropic Re-entry
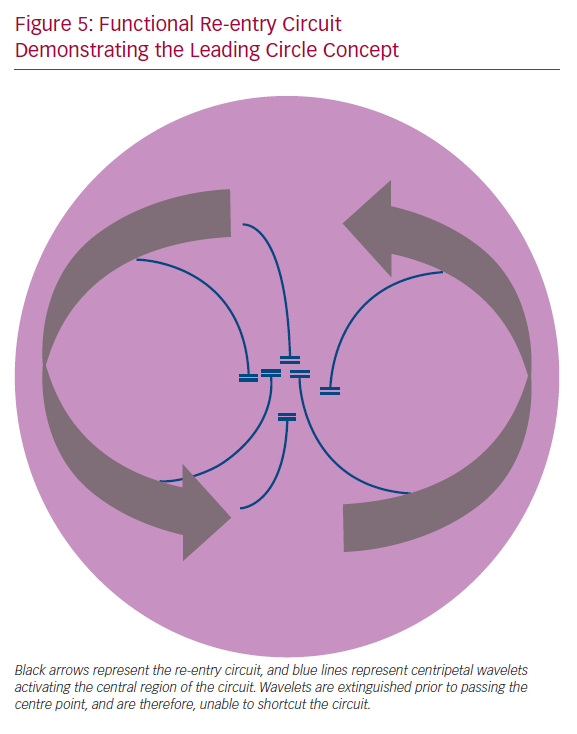
In addition to UCB, re-entrant circuits characteristically require a region of inexcitability for the re-entrant wave to circumnavigate. This may take the form of a fixed anatomic obstacle to conduction, such as a valve or blood vessel, or if occurring in the absence of such an obstacle, can be termed ‘functional re-entry’. Functional re-entrant circuits have been observed experimentally.70 Originally, this was explained by the ‘leading circle’ concept, whereby the central region of the re-entry circuit is activated by multiple wavelets branching from the main re-entry circuit (Figure 5). Experimental data indicate that this functional re-entry may arise due to heterogeneity in the refractory properties of the tissue, but may also arise in the absence of marked differences in refractoriness, exclusively due to anisotropic tissue properties of conduction.28,71 Modelling studies have suggested that functional re-entry within the pulmonary veins may underlie rapid activation that results in paroxysms of AF, which in the present study was dependent on heterogeneous and anisotropic conduction within the pulmonary veins.72,73 CV anisotropy therefore represents a distinct mechanism underlying sustained functional re-entry. In an experimental setting, the re-entrant path of these anisotropic re-entrant circuits is closely related to myocyte orientation.74
Spiral Wavebreak
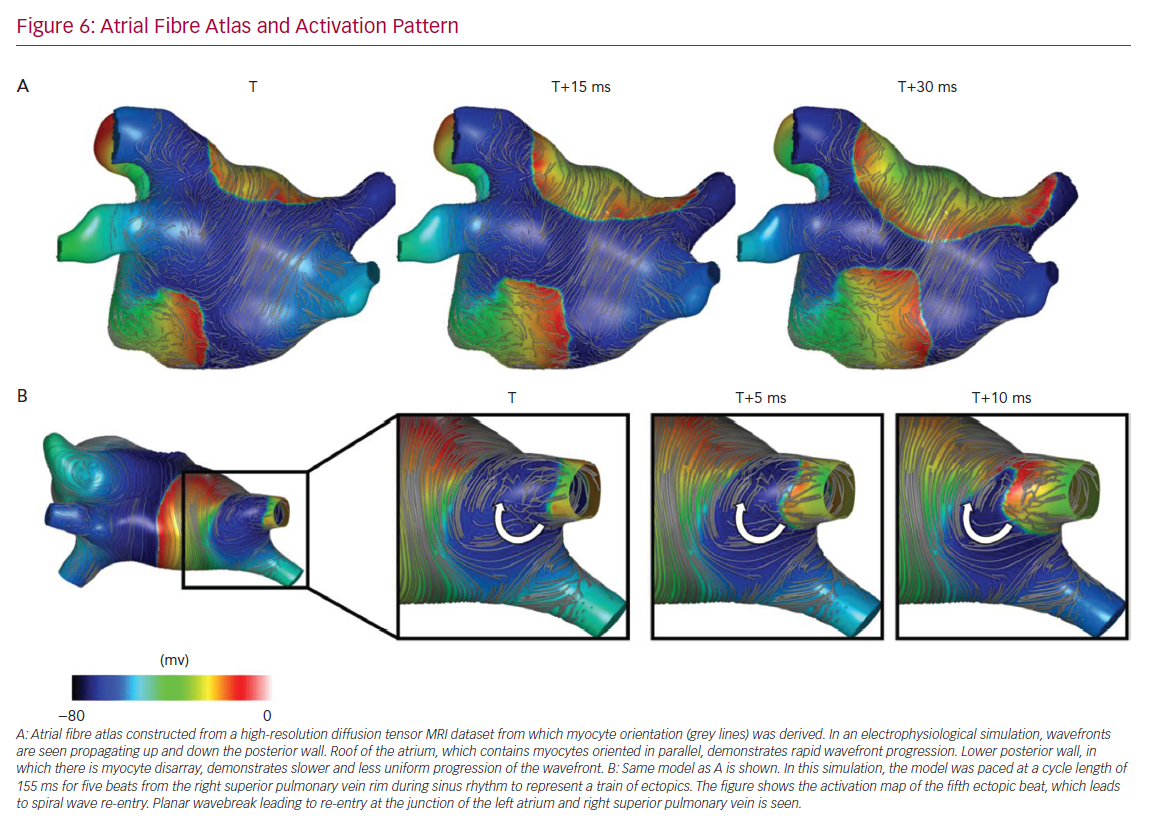
Junctions in myocyte orientation are observed at typical places in the atria (an example of myocardial arrangement is shown in Figure 6), and represent locations where the propagation of a spreading wavefront is subject to changes in CV and anisotropy.75
In 3D tissue, spiral waves manifest as a scroll wave or vortex. The break-up of a scroll wave is a potential mechanism underpinning a transition from a tachycardia to fibrillation. The centre of the scroll or filament forms a line through the centre of the scroll analogous to the phase singularity in 2D spiral waves. The relationship between CV anisotropy and filaments has predominantly been investigated using mathematical models. Mathematical formulations and simulation studies have shown that the motion of scroll wave filaments is determined by local anisotropy.76 Simulations have predicted that tissue anisotropy promotes filament motion towards the apex in the ventricles.77 In addition, simulations have shown that spatially varying anisotropy promotes both bending of the filament and slow wave speed due to curvature, which can contribute to wavebreak.78 The stability and location of scroll waves in cardiac tissue are highly dependent on tissue CV anisotropy.
Spiral waves (in 2D) or scroll waves (in 3D) are examples of functional re-entrant circuits that are thought to be involved in the mechanisms underlying AF and VF, and have been observed more frequently in ventricular locations at abrupt junctions of myocyte orientations.79–82 Furthermore, junctions where myocytes meet in different directions are regions that favour the development of UCB, as well as promoting wavebreak, which is observed in fibrillation and may be a mechanism by which fibrillation is sustained.83,84 These considerations illustrate the importance of myocyte orientation and anisotropic conduction properties in the development and maintenance of functional myocardial re-entrant circuits, and suggest mechanisms that may promote fibrillation.
Ectopic Foci
In addition to promoting re-entry, anisotropy of conduction has been implicated in arrhythmia initiation through the propagation of wavefronts from ectopic foci of depolarisation. In simulation studies, a critical degree of cellular uncoupling related anisotropy, for example, that which may be seen with myocardial fibrosis, will permit propagation of an ectopic wavefront, which would otherwise extinguish.85 Ectopic foci are well known to play a crucial role in the initiation of clinical arrhythmias, including AF.73
Outstanding Questions
CV anisotropy is a characteristic of myocardial tissue. The speed of conduction is reliably greatest in the direction parallel to the longitudinal orientation of myocytes. As such, there is an intimate duality between conduction anisotropy and myocyte orientation. Both CV and ARCV change following pathological tissue remodelling and are associated with pro-arrhythmic states. Uncertainty remains regarding the magnitude of the change, which is likely to depend on the location and pathological process considered. There are compelling explanations and experimental evidence demonstrating mechanisms by which changes in anisotropy may promote re-entry. Some of the discrepancies in the available evidence relate to the experimental models used. Most data have been collected in experimental models due to the difficulty of assessing both myocyte orientation and anisotropy in vivo, and as such, there is residual uncertainty as to the translatability of specific experimental results to human physiology. Recently, an estimation of human atrial myocyte orientation in ex vivo hearts has been made, establishing that through the use of diffusion-tensor MRI (DT-MRI), myocyte orientation of both epicardial and endocardial atrial tissue layers may be estimated, which was previously only possible through direct tissue examination.86 Furthermore, recent reports have successfully demonstrated that DT-MRI may be reproducibly applied in vivo. It has been used to demonstrate myocyte disarray in hypertrophic cardiomyopathy patients and has been correlated with the incidence of ventricular arrhythmias.87,88 In this case, decreased structural anisotropy (when averaged over the imaged voxel volume), representing myocyte disarray, was associated with an increased incidence of ventricular arrhythmias. Importantly, the identification of decreased averaged structural anisotropy must be distinguished from the conduction characteristics of myocardium in hypertrophic cardiomyopathy, which remains highly anisotropic in fibrotic areas.25 It is hoped that DT-MRI application to other pathologies associated with myocyte disarray and arrhythmias will follow and further define the structural characteristics of the myocardium in pathological conditions. An alternative approach to in vivo imaging has been proposed to establish both myocyte orientation and ARCV in the human atrium using activation patterns during pacing from multiple sites in a process designed to overcome the challenges of wavefront curvature and propagation of wavefronts non-parallel or perpendicular to myocyte orientation (Figure 6).89 If validated, such a tool may facilitate the establishment of endocardial atrial myocyte orientation variability, as well as the key functional aspect of change in ARCV under different pathological conditions. Further data regarding typical myocyte orientation and ARCV with different pathological states, either through structural imaging or a functional assessment, may contribute to a greater understanding of arrhythmia mechanisms responsible for re-entrant arrhythmias, including AF. Furthermore, such information may be helpful to predict, and possibly guide, the effect of specific anti-arrhythmic medication in the context of defined changes in substrate behaviour. Such an assessment may also allow the identification of regions, for example, those with markedly increased anisotropic conduction, that could be targeted for ablative therapy. Accurate information regarding myocyte orientation and CV anisotropy in an individual atrium would allow a more thorough assessment of the relationship between imaging features (e.g. late-gadolinium enhancement MRI) and functional electrophysiological behaviour, which has rarely been incorporated into previous assessments.90 It would also allow the parameterisation of patient-specific computational models that may be used to guide individualised precision therapy.
Conclusion
Myocyte orientation and CV anisotropy are fundamental tissue properties that are important determinants of susceptibility to arrhythmia. Myocyte orientation determines the direction of maximal CV. The ratio between CV in the direction of maximal and minimal CV, the ARCV, represents a determinant of tissue susceptibility to re-entry through the promotion of UCB. In addition, anisotropic conduction itself represents a mechanism that may explain the existence of functional re-entry within cardiac tissue. Tools for establishing myocyte orientation and conduction anisotropy in vivo may address outstanding questions regarding the magnitude and mechanism underlying changes in conduction anisotropy in pro-arrhythmic pathologies.
Clinical Perspective
- Tissue anisotropy is dependent on myocyte orientation.
- Anisotropic myocardial conduction is enhanced in pathological states, which may contribute to arrhythmogenesis, through the promotion of abnormal focal activation, as well as functional re-entrant arrhythmias.
- If identifiable during clinical procedures, areas of enhanced anisotropic conduction may represent novel targets in which ablative therapy could be trialled if demonstrated to promote fibrillation.