










In AF patients, the initiation and progression of AF depend on the underlying arrhythmogenic substrates, genetics and concomitant risk factors, which may impact the clinical presentation of the arrhythmia and manifestation in different characteristic AF patterns. Current research projects focus on the development of several diagnostic techniques for the assessment of arrhythmogenic substrates in patients with AF (diagnostic translation). Additionally, multidisciplinary teams (collaborative translation) try to describe genetic mechanisms, molecular pathways, electrophysiological characteristics and concomitant risk factors with the goal to provide a more mechanism-tailored classification of AF with the potential to improve AF treatment in subgroups of patients.
However, despite emerging evidence of the prognostic value of several new techniques and the potential utility to guide AF management, most of these strategies have not yet been implemented in the clinic. In this article, current approaches for AF substrate characterisation, analysis of genes potentially involved in AF and strategies for AF risk factor assessment are summarised. We discuss challenges and obstacles to clinical translation and implementation into clinical practice.
AF Pathogenesis
Large-animal models of AF have been of great importance in understanding the pathogenesis of AF. Seminal early studies demonstrated that AF is a self-perpetuating arrhythmia because it causes proarrhythmic remodelling of the atria, both electrical and structural.1 Electrical remodelling is a relatively fast process, complete within 1–2 days of AF onset.2 It is characterised by a shortening of the atrial action potential and a corresponding reduction in the atrial refractory period, allowing higher fibrillation frequencies. As such, it is a pivotal factor in the early stabilisation of AF, when anti-arrhythmic drugs are still effective. Structural remodelling, which is thought to be responsible for the further progression of AF, develops over a much slower time course of months to years and is typified by myocyte hypertrophy, deposition of fibrous tissue and expansion of adipocytes.3,4
Atrial electrical and structural remodelling occur not only as a result of AF, but also due to factors increasing the risk for AF. Atrial remodelling has been studied in large animal models of, e.g. congestive heart failure (CHF), valvular disease, hypertension, sleep apnoea and obesity.5–9 In a goat model of ‘lone AF’, increased complexity of fibrillatory conduction and a loss of efficacy of anti-arrhythmic drugs was associated with the development of endomysial fibrosis, i.e. increased thickness of collagen septa between myocytes within bundles (also called reactive or interstitial fibrosis) and typically occurring in the absence of myocyte death.10,11 In contrast, a dog model of heart failure did show myocyte death and a corresponding pattern of replacement fibrosis.12 In a direct comparison with a dog model of lone AF, the CHF model showed relatively simple fibrillation patterns and a higher efficacy of pharmacological cardioversion.13 This strongly suggests that the exact pattern of fibrosis, rather than the overall amount of fibrous tissue, determines AF complexity and stability, in a manner reminiscent of the relationship between fibrosis pattern and conduction abnormalities observed in the ventricle.14,15 In aggregate, evidence from diverging studies illustrates the complex interplay between anatomy, fibre architecture, patterns of fibrosis and other types of structural remodelling and their effect on AF pattern. For example, larger areas of (replacement) fibrosis may serve as an ‘anchor point’ for spiral waves and other macroreentrant patterns, whereas endomysial fibrosis with loss of side-to-side connections may precipitate microreentry or ‘zig-zag’ conduction even in relatively small tissue areas.16–18
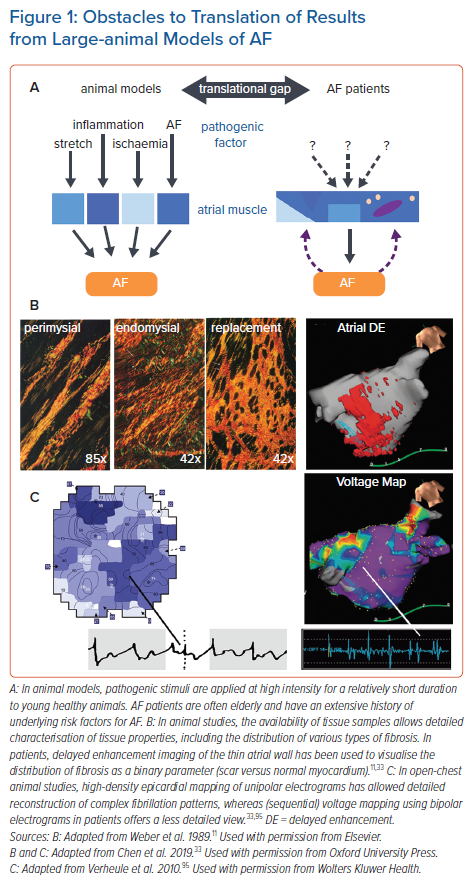
Obstacles to Clinical Translation Related to the Description of AF Pathogenesis
In contrast to the controlled circumstances and limited duration of pathogenic stimuli in animal models, AF patients often have several risk factors (including ageing) that may have progressed over a prolonged time span, contributing to the ‘translational gap’ between insights from animal model and clinical practice (Figure 1).19 One important aspect of AF pathogenesis in patients that has not been replicated in animal models is the occurrence of spontaneous AF episodes originating in the pulmonary vein region. Whether this results from differences in anatomy, tissue architecture, or pathogenesis, as mentioned above, is currently unclear, but the lack of large animal models showing spontaneous AF paroxysms limits our understanding of the underlying mechanisms of this important manifestation of AF in patients.
The ‘translational gap’ between animal studies on AF pathogenesis and patient studies extends to diverging methods used for electrophysiological mapping of the AF substrate. Most mapping studies in animals were performed in open chest situations with epicardial arrays of unipolar electrodes. Although some mapping studies in patients implemented similar techniques during cardiac surgery, most were performed using either bipolar endocardial electrodes or non-contact mapping.20,21 Unipolar electrograms allow more precise determination of local activation times and provide information on the degree of fractionation without sensitivity to the direction of propagation.22,23 Moreover, epicardial access provides higher spatiotemporal resolution and, therefore, more accuracy in the reconstruction of the complex fibrillation patterns that are associated with AF progression.
In animal models, tissue samples for the detailed analysis of structural remodelling can be obtained under controlled conditions at different time points. In general, studies in human atrial samples remain essential to study the consequences of clinically relevant disease-related remodelling in patients. For example, recent work identified the proarrhythmic molecular and cellular mechanisms contributing to AF in patients with heart failure and sleep apnoea.24–26 However, human atrial samples are only available from patients undergoing cardiac surgery, who likely have a risk factor profile that is dominated by ischaemic heart disease and/or valve disease and may not be representative for all AF patients. Furthermore, atrial samples are often restricted to right or left atrial appendages, which may undergo remodelling distinct from regions that are considered more relevant for AF maintenance (e.g. the pulmonary veins and the left-atrial free wall). Nonetheless, right-atrial sources for AF are increasingly reported.27,28 Most importantly, human atrial samples can only provide a single snapshot of the cumulative effect of all AF risk factors, ignoring their complex temporal dynamics.29
As an alternative to histological analysis, several surrogate parameters have been developed for imaging of atrial tissue characteristics. These methods have an intrinsically lower resolution and are limited by the thinness of the atrial wall. Thus, imaging of fatty deposits does not allow the distinction of epicardial fat from intramyocardial fatty infiltrates.30 As a measure for fibrosis, imaging of regions with delayed enhancement of gadolinium has been used increasingly in recent years.31,32 Although this method has been validated histologically for large infarct scars in the thick ventricular wall, direct histological validation of delayed enhancement imaging in the atrium has been very limited.33 Therefore, the exact type and distribution of fibrous tissue (i.e. reactive versus replacement fibrosis) detected using this technique remains uncertain. Nevertheless, areas with delayed enhancement have been correlated to regions with low-voltage electrograms, and are predictive of clinical outcome.34,35 Most notably, patient-specific mathematical models incorporating information from delayed enhancement imaging have been successfully used to guide ablation strategies.35
Another strategy might be collection of blood biomarkers, representing different pathological pathways to evaluate the progression of atrial cardiomyopathy.36 Although not specific to atrial myocardial disease, various biomarkers characterising myocyte injury, inflammation and fibrosis have been linked to the occurrence and outcomes in AF. From an electrophysiological and pathophysiological point of view, biomarkers that characterise the atrial substrate may be more indicative of AF burden rather than the seemingly random time point at which AF is first documented.37
AF Genetics
Over the previous few decades, tremendous strides were made in elucidating the genetic underpinnings of AF.38 Family-based methods, primarily linkage and fine-mapping successfully detected numerous loci leading to Mendelian forms of AF. At least eighteen forms of monogenetic AF are described, including those resulting from mutations in genes that code for potassium voltage gated channels (KCNQ1, KCNE2, KCNA5 and KCNJ2) and sodium voltage-gated channel subunits (SCN1B, SCN2B, SCN3B, SCN4B and SCN5A), natriuretic peptides (NPPA), gap junctions (GJA5), nucleoporins (NUP155), ATP binding cassette transporters (ABCC9) and myosin light chains (MYL4).39–41 Causal genes have not been located for a number of other implicated regions.42 These findings generally involve rare, highly penetrant mutations with large effect sizes, but small population level risk.
The mutations identified in family-based studies demonstrate the fundamental importance of two ion handling groups of proteins that can influence AF aetiology through a variety of related molecular mechanisms. The potassium handling family of genes, for example, is involved in multiple facets of potassium induced current flows, particularly delayed rectifier outward currents (KCNQ1, KCNE2, KCNA5) and inward rectifier currents (KCNJ2, ABCC9).43,44 Mutations in β subunits of the voltage-gated sodium channels (SCN1B, SCN2B, SCN3B, SCN4B) have often been associated with decreases in sodium current density and/or late sodium currents, while those in α subunits, including SCN5A, lead to decreased channel expression and inactivation abnormalities and are associated with a considerable degree of phenotypic variability.45,46 Variants in the genes coding for both protein families alter cardiac action potentials.
Genome-wide association studies (GWAS) of AF, by contrast, delineated numerous common polymorphisms with generally small effect sizes, but affecting larger segments of the population in individuals of European ancestry and others.47–55 These GWAS have been supplemented by whole exome sequencing and exome-chip analyses that have identified additional pertinent loci.56,57
Obstacles to Clinical Translation Related to AF Genetics
Despite the success of these studies in locating a large number of loci associated with AF and AF endophenotypes, substantial progress in translating this information into clinically relevant knowledge has largely not been realised to date. In familial settings, characterising mutations in currently unaffected individuals may lead to the prospective identification of cases. These data have also proven useful in terms of helping to characterise mechanisms that contribute to the development of AF. Nevertheless, given the rarity of these mutations, family-based data typically finds limited applicability in the general population.
GWAS, in contrast, has identified many common genetic AF susceptibility variants throughout the genome. Polygenic risk scores derived from these can be used to successfully predict a person’s risk of developing AF.58 The authors of that study suggest that, “the identification of individuals at high risk should facilitate the design of efficient natural-history studies to discover early markers of disease onset and clinical trials to test prevention strategies.”
However, direct translation of GWAS data, outside of this domain has been complicated by a number of factors. These include the difficulty in determining the causal variant and the gene it affects, at most loci due to the effects of linkage disequilibrium, the fact that most GWAS variants are intergenic and the cost, in terms of both money and manpower, of following up on the large number of GWAS findings.
These problems are exacerbated by the typically small effect sizes of these variants, which complicates the task of prioritisation for follow-up. However, it is worth noting that variants with small effects may implicate genes which may be potent therapeutic targets. An excellent example is the 3-hydroxy-3-methylglutaryl-coA reductase (HMGCR) gene. Common GWAS variants in this gene are associated with LDL cholesterol levels, albeit with a comparatively modest effect size of ~0.06 mmol/l.59 The statin drugs, which act by inhibiting the protein product of the HMGCR gene, on the other hand, have large effects on serum LDL.60 This example – and others – suggests that validation and functional characterisation of GWAS findings may provide both mechanistic and clinically actionable insights into AF.
Moreover, the value of GWAS, as well as genome- and exome-wide DNA sequencing studies, can be further exploited in several ways. The first is to use more precisely defined clinical phenotypes to discern genetic variants that may be associated with etiologically distinct forms of AF. The second is to perform genome-wide studies on novel endophenotypes, such as tissue characteristics (i.e. fibrosis, capillary rarefaction and fatty infiltration), sequential voltage mapping, unexplored ECG parameters, etc. Although it may not be feasible to ascertain sample sizes on the same scale as previously studied traits, these studies may yield findings that help to elucidate the genetic complexities of AF.
Other forms of analysis may also shed light on AF. An important branch of emerging research examines the transcriptomics involved, either causally or in response to AF. Previous studies have suggested that transcriptional profiles are associated with AF; however, these studies have typically been hampered by the use of microarrays, in lieu of next-generation RNA sequencing, small sample sizes, limited analysis of selected transcripts and lack of replication.61 On-going studies, involving large numbers of atrial biopsies and state-of-the-art RNA sequencing methods, will allow unprecedented opportunities to evaluate the transcriptional landscape of AF, related endophenotypes (such as PR interval and P wave characteristics), markers of atrial remodelling and AF risk factors. As evidence continues to accumulate implicating the crucial role of non-coding RNA (including long non-coding RNA, microRNA, circular RNA, small nucleolar RNA and other species) on transcriptional regulation, RNA sequencing should yield considerable insight into not just genes involved in AF but the mechanisms of their control. These analyses will provide valuable information on the causes and consequences of AF, AF aetiology and AF biomarkers.
RNA sequencing data will also provide valuable clues to the role of AF-associated genetic variants.62 Moreover, the integration of genomic and transcriptomic data will allow for more precise classification of AF patients – a goal considered crucial in the field.63 The application of machine-learning methods, such as random forests, will also yield more precise phenotypes for further genomic and transcriptomic discovery (Figure 2).
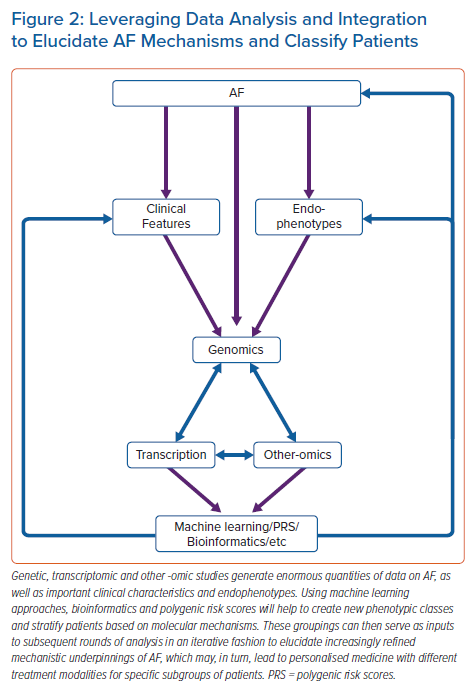
AF Risk Factors
Several cardiovascular risk factors contribute to AF initiation and progression (see the AF Pathogenesis section). In the current guidelines of the European Society of Cardiology for AF diagnosis and management, the importance of identification and management of established AF-promoting risk factors and unhealthy lifestyle is addressed and clearly recommended.63 In addition to non-modifiable factors such as advancing age and sex, hypertension, obesity, excessive alcohol consumption, sleep apnoea and endurance exercise, in particular, are separately mentioned.64 For all these modifiable AF risk factors, meta-analyses of observational studies or randomised intervention studies support the recommendations for clinical management of these conditions.65–69 Aggressive management of AF risk factors combined with lifestyle changes has been shown to control sinus rhythm in AF patients.65 Despite these described positive responses to risk factor management programmes in AF, other studies could not confirm these beneficial effects, suggesting, that there are potentially non-weight responsive forms of AF in some patients.70–73
Previous work showed that idiopathic AF patients develop cardiovascular disease more often, at a younger age and with a more severe disease profile compared to healthy sinus rhythm controls, suggesting the presence of subclinical comorbidities.74 Similarly, in a recent retrospective study of young AF patients (mean age 46 years), only 11% of patients were free of AF risk factors or comorbidities, whereas 44% had hypertension and 25% had a family history of AF.75
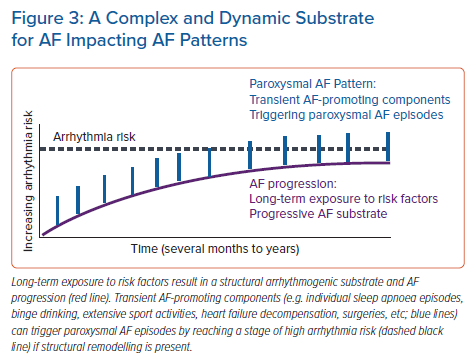
Despite the available evidence and the recommendations in current AF guidelines, the optimal approach to assess AF risk factors and specific lifestyle components remains unclear. In clinical practice, the presence of AF risk factors is generally considered a binary variable. However, AF risk factors (with the exception of the genetic component) may be dynamic, which results in a temporally variable exposure of the organism to arrhythmogenic conditions that may contribute to variability in the frequency, duration and timing of paroxysmal AF episodes.76 In addition to the structural remodelling process, which generally develops and progresses slowly, the exposure to variable AF risk factors may also contribute to temporal patterns of paroxysmal AF episodes by transient acute arrhythmogenic mechanisms.77 This may result in fluctuations in triggers and components of the substrate that can be rapidly modified such as post-translational modifications of proteins or autonomic nervous system activation, etc.19 The concept of a dynamic substrate for AF is summarised in Figure 3.
Each AF risk factor has different dynamic components with distinct time courses. Advancing age is a slowly progressive AF-risk factor. Long-term untreated hypertension also has a strong progressive component. Frequent alcohol intake is an important risk factor for new-onset AF. Furthermore, binge drinking, can also trigger paroxysmal AF episodes, known as the ‘holiday heart syndrome’.78
Intense activity, sleep apnoea, excessive alcohol intake and surgery have major transient AF-promoting components that are due to modulation of systemic regulators such as activation of the autonomic nervous system in the case of exercise, intrathoracic pressure changes in sleep apnoea or inflammation following cardiac surgery. 8,79,80 These transient effects are partially or fully reversible and are modulated by variability in risk-factor severity.
The VARIOSA-AF study is an example how transient AF-promoting factors can influence AF patterns: there is considerable night-to-night variability in sleep apnoea severity, which impacts AF risk associated with sleep apnoea severity in the preceding night.81,82 Additionally, over time these variable risk factors may also promote progressive structural remodelling processes. In rats, cumulative exposure to transient sleep apnoea-related conditions resulted in AF-substrates and was associated with increased AF-susceptibility.83 Of note, AF risk is not just determined by risk factors with clinical manifestation but also by variability in sub-clinical risk factors. In AF-patients without overt risk factors, the inter-visit variability of metabolic parameters showed a close association with AF risk.84 Additionally, the combination of risk factors may be important as well. For example, rats with spontaneous hypertension and obesity show more pronounced structural remodelling and AF susceptibility than lean hypertensive rats.85
A binary assessment of AF risk factors, as currently practised clinically, may not adequately reflect the contribution of the respective risk factor on AF mechanisms and progression. Under circumstances of variable risk factors, the development of the AF substrate will depend critically on the cumulative exposure to the arrhythmogenic conditions (the risk factor burden represents the time exposed to a particular risk factor).
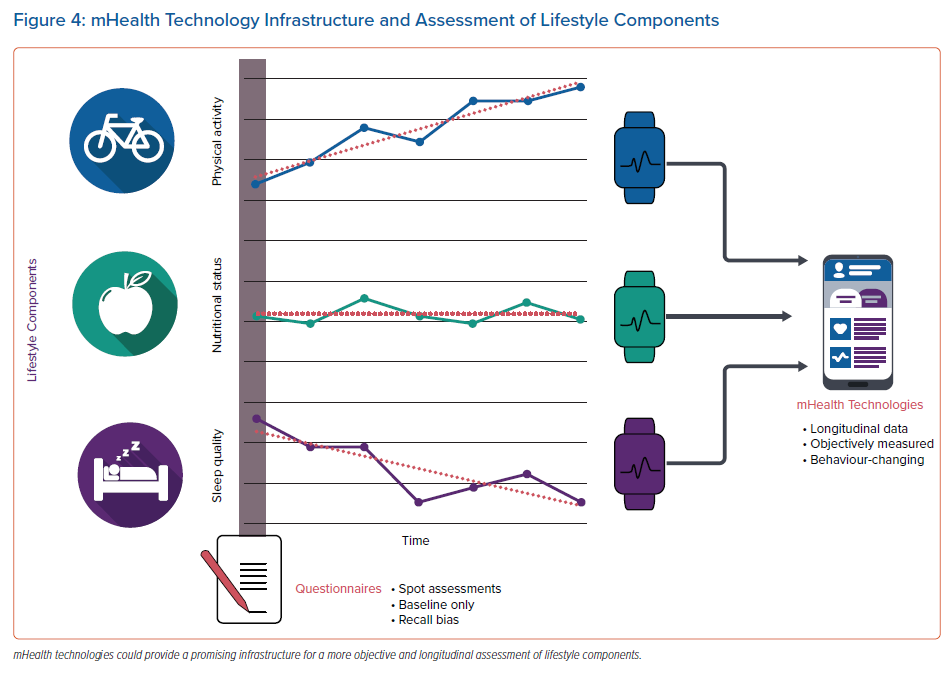
Despite convincing evidence of the need for risk factor management in AF patients, it remains unclear how best to assess substrates and risk factors and guide treatment, risk factor management and lifestyle modification.64 Established risk factors are often assessed only once in a structured way at the time point when patients present for the first time in the AF-clinic (spot-assessment of risk factors). However, several AF risk factors may show a high visit-to-visit or even day-to-day variability and lifestyle components, such as physical activity, diet and sleep behaviours, may be variable over time. Therefore, clinically relevant risk factors will be missed if the assessment is only performed once. Importantly, this variability does not just complicate the detection of AF risk factors but may also have a prognostic implication. High visit-to-visit variability in risk factors is associated with increased risk of incident new-onset AF, worse cardiovascular outcome and increased mortality.86 Additionally, lifestyle components are often self-reported or assessed once by questionnaires. Lifestyle, though, may vary (such as seasonal variation), again resulting in a dynamic exposure to lifestyle-related conditions which may critically impact the timepoint and extent of incident AF episodes (Figure 4).
Risk factor assessment, therefore, requires a longitudinal and remote structured monitoring infrastructure. Additionally, longitudinal documentation of risk factors during a modification programme may allow monitoring of the response to the intervention and adaptation and guidance, as required, to optimise the results. Recent technological advances have considerably expanded the options for non-invasive, longitudinal assessment of AF and potential underlying risk factors. Implantable loop recorders and a variety of wearables enable longitudinal or (near) continuous AF monitoring, making it possible to directly link specific clinical conditions to the occurrence of AF. Furthermore, longitudinal risk factor monitoring is becoming increasingly feasible. For example, some pacemakers can perform continuous monitoring of sleep disordered breathing, and implantable pulmonary arterial pressure monitors provide haemodynamic information that is used in remote monitoring of HF patients.74,87 Finally, some blood-based biomarkers have been associated with AF.37 Repeated biomarker collection could potentially provide information about progressive atrial remodelling, although at present there are no biomarkers that specifically reflect individual AF mechanisms.
mHealth apps and smart technologies, such as activity trackers, Bluetooth-linked balances, blood pressure devices and apps to assess diet, may allow a widespread and affordable infrastructure for longitudinal risk factor identification and monitoring (Figure 4).88 Several mHealth applications are available for the management of AF.89 In addition to the assessment of risk factors, mHealth infrastructures and apps can also be helpful in applying dedicated in-app coaching to improve lifestyle and control risk factors by behavioural changes.90–93
Obstacles to Clinical Translation Related to AF Risk Factors
Implementation of infrastructure for longitudinal assessment of variables such as risk factors, lifestyle components, or rate and rhythm information require adaptation of existing care coordination and clinical pathways.94 An important element for embedding mHealth in clinical practice is the accessibility of the recordings by other healthcare professionals. For this, a connection with the patients’ electronic healthcare record is crucial. Additionally, many available devices and apps lack scientific validation and are written at excessively high reading-grade levels challenging users with limited health literacy.
Although technologies for longitudinal monitoring of lifestyle components and AF risk factors are available and may represent an interesting tool in future research projects, legal considerations and missing reimbursement models are still blocking wide implementation in existing clinical pathways. A multidisciplinary effort by regulatory agencies, healthcare organisations and app sellers is required to improve relevance, scientific validity and readability of AF apps for AF patients. Additionally, discussions with insurance companies about reimbursement of mHealth infrastructures and with different stakeholders to agree on security and privacy regulations are initiated in different countries.
Bridging the Translation Gap
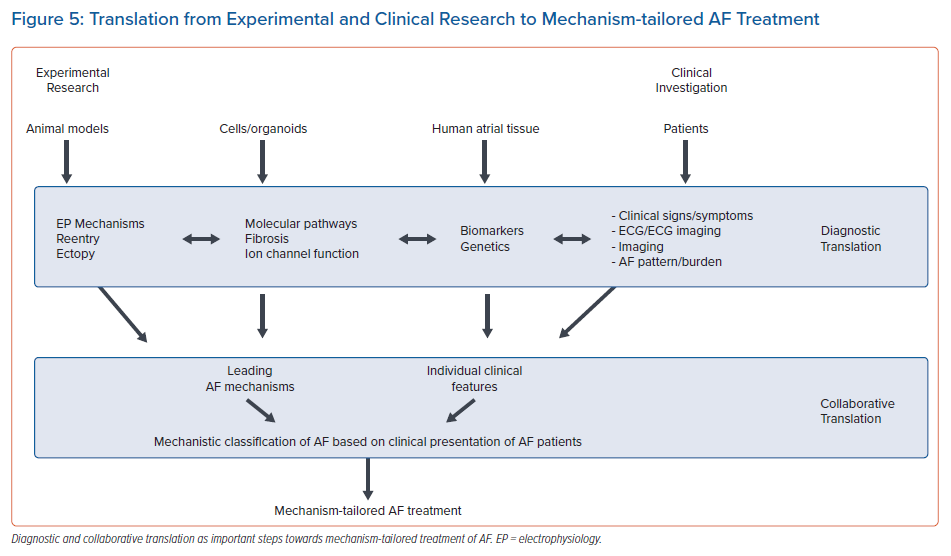
Successful translation to develop a mechanism-tailored classification of AF with the potential to improve treatment in subgroups of patients will require translational research approaches on various levels (Figure 5). Diagnostic translation involves development of clinical diagnostic tools allowing the identification of mechanisms of AF. Examples are biomarkers for fibrosis or non-invasive electrophysiological markers of the degree of electrophysiological changes in the atria. Additionally, several large ongoing studies are attempting to establish associations between the clinical profile of AF patients derived from deep phenotyping with leading AF mechanisms identified on the tissue level. This approach requires multidisciplinary teams studying genetic mechanisms, molecular pathways and electrophysiological characteristics on the tissue level in clinically well characterised patients (collaborative translation). Such approaches recently became feasible with advances in non-invasive or minimally invasive characterisation of AF patients (non-invasive electrophysiology, longitudinal risk factor assessment, AF-burden and patterns using mHealth technologies, wearables or implantable loop recorders) and the development of high throughput histological techniques and -omics approaches, such as RNA sequencing.
The development of a new mechanism-tailored classification AF will be essentially an iterative process, where particular investigational steps informed by initial outcomes are repeated to refine the classification. The application of machine learning methods will result in more precise phenotypes for further clinical investigations. This will, in turn, lead to a more exact AF taxonomy. Both diagnostic and collaborative translation aim at the development of a mechanism-based subclassification of AF potentially allowing for better treatment of subclasses of AF patients. Supplementing these efforts with other current technological innovations, such as epigenetic sequencing, proteomics, metabolomics and microbiomics, will further the goal of developing mechanistic insights into AF pathology. The implementation of these methods, especially in conjunction with genomic and transcriptomic data, will further aid efforts to define molecular sub-types of AF aetiology and pathology.
Clinical Perspective
- The best way how to assess substrates, genetics and risk factors in patients with AF remains unclear.
- A successful translation of research focussing on atrial arrhythmogenic mechanisms has the potential to provide a mechanism-tailored classification and to support personalised treatment approaches in patients with AF.
- The clinical uptake and clinical implementation of new diagnostic techniques and treatment strategies require translational research approaches.