







Atrial fibrillation (AF) is a highly prevalent arrhythmia that represents an important burden on healthcare systems. The presence of AF is associated with an increased risk of conditions such stroke, heart failure and dementia. Further, AF is associated with increased mortality. Over the past half century, significant advances have been made in understanding the pathobiology of AF. Important among these have been the demonstration that AF is a heritable disease and the identification of genetic variants underlying AF. The following review provides an overview of research in AF genetics followed by a discussion on the potential applicability of AF genetics research to clinical practice. The literature review was conducted in the PubMed database between January 1940 and January 2014.
Historical Perspective
The first reports suggesting a genetic basis of AF emerged in the 1940s when Wolff described an AF pedigree with a number of affected siblings.1 In the ensuing decades, multiple rare AF pedigrees with monogenic patterns of inheritance were described. However, it was not until 2003 that the first mutation in an AF family was reported. Using classic genetic techniques, such as linkage analysis and candidate gene screening, Chen et al. identified a potassium channel mutation in a four-generation pedigree with autosomal dominant AF. Over the next few years, much of the research in AF genetics focused on familial forms of the arrhythmia and led to the identification of additional genetic mutations (discussed in more detail in the next section).2–31
Around the same time as the first reports of causative mutations in AF pedigrees, epidemiological studies began to emerge suggesting that the form of AF encountered in everyday clinical practice also has a significant genetic component. The first large population-based study to report familial clustering of AF came from investigators at the Framingham Heart Study. They reported that more than a third of AF cases in the general population have a relative with the arrhythmia.32 Two subsequent studies from Iceland and the US also reported familial aggregation in cohorts of lone AF patients.33,34 More recently, a Danish study involving more than 9,000 lone AF patients demonstrated a strong familial component to the arrhythmia.35
While familial AF is caused by single gene mutations, the form of AF encountered in everyday clinical practice is likely to be a more complex trait, which is caused by multiple genetic variants interacting with environmental factors. The identification of the genetic architecture underlying the common form of AF has represented a challenging task. Candidate gene association studies have attempted to identify common variants underlying AF with limited success.
The recent emergence of next-generation sequencing (NGS) technology has enhanced the ability of researchers to identify genetic variants underlying complex traits. Since 2007, genome-wide association studies (GWAS) have used NGS technology to identify multiple variants underlying AF. NGS technology has also led to the development of exome sequencing and whole genome sequencing, which allow simultaneous sequencing of the entire protein coding region or the whole genome, respectively. These are promising techniques for the identification of causative variants in AF pedigrees as well as AF populations. However, as yet, they have not been widely applied to AF genetics research.
Rare Mutations in Familial Atrial Fibrillation
Linkage analysis and candidate-gene sequencing have identified multiple mutations in monogenic AF families and isolated AF cases.2–31 Linkage analysis involves performing genotyping of markers distributed throughout the genome and investigating the transmission of these markers with disease within a pedigree.36 Markers that transmit closely with disease lie in proximity to the disease-causing mutation. Therefore, identifying a series of markers that transmit closely with disease narrows the search space for the disease-causing variant to a defined subsegment of the genome. The genes within the sub-segment can then be screened to identify a causative mutation. However, conventional genotyping techniques are often time consuming and demanding.
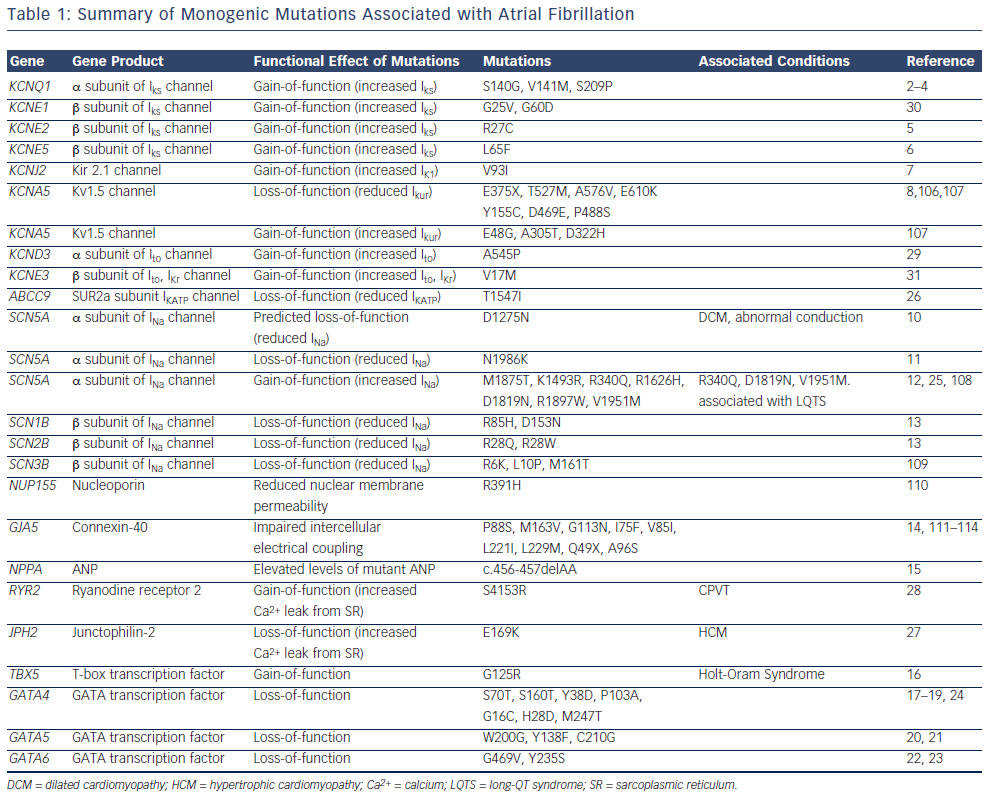
The majority of reported mutations for familial forms of AF are located in genes that encode ion channel subunits (see Figure 1 and Table 1). The identification of these mutations led researchers to question whether single-gene mutations in ion channel genes also contribute to the heritability of the common form of AF in the general population. Early reports from candidate-gene screening studies suggest that these mutations are not prevalent in the general population.11,13,30,37–41 Of note, however, the genes identified in AF pedigrees are significantly more likely to harbour rare variants in cohorts of lone AF patients compared with control populations.42
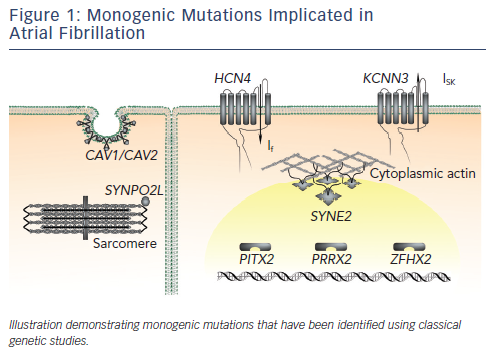
While the mutations identified in monogenic AF pedigrees are rare, their identification has provided interesting insights into the pathogenic basis of AF. Both gain-of-function and loss-of-function mutations in genes encoding potassium and sodium channel subunits have been reported to underlie familial AF.2–9,11–13,25,26,29–31,43 Gain-offunction potassium channel mutations are predicted to influence AF by shortening the atrial effective refractory period, an effect that would be predicted to promote atrial re-entry.44 Loss-of-function potassium channel mutations are predicted to promote triggered activity in the atrium, which is also an important contributor to the genesis of AF.8 Gain-of-function sodium channel mutations are predicted to promote triggered activity.25 Loss-of-function sodium channel mutations are predicted to shorten the wavelength of an impulse circulating around a re-entry circuit.45 The potential mechanistic links between non-ion channel mutations and AF pathogenesis are less clearly understood.
Common Genetic Variants and Atrial Fibrillation in the General Population
Identifying the genetic basis of the common form of AF in the general population is a more challenging task. Genetic association studies are valuable tools in this context. In contrast to family-based studies, which investigate co-segregation of genetic variants, population-based association studies investigate the co-occurrence of genetic variants in affected individuals. It is important to emphasise that in contrast to family-based studies, in which the reported mutations have large effect sizes and are directly responsible for causing the trait, association studies identify variants with small effect sizes that confer an increased risk of disease. Functional validation of the role of these variants in disease pathogenesis is typically more difficult.
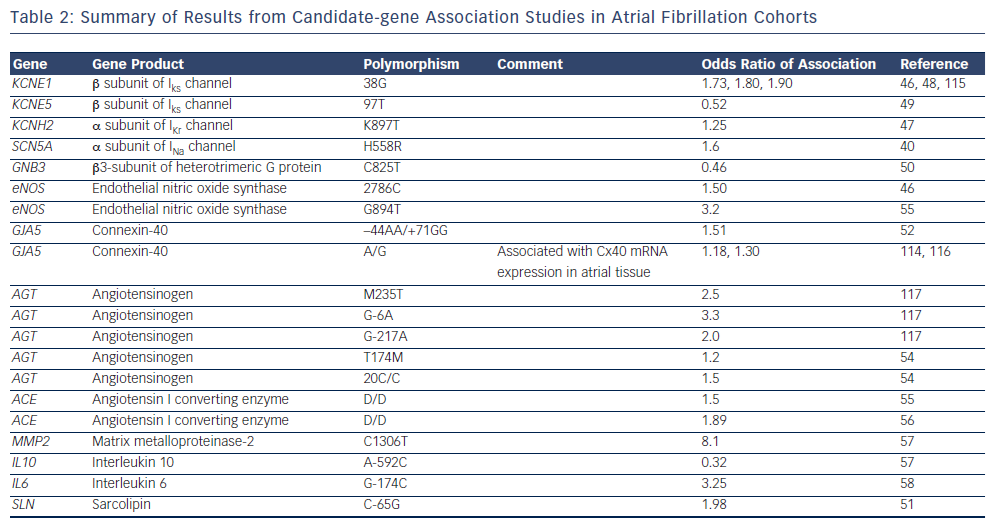
The majority of early association studies in AF were candidategene association studies.40,46–58 Candidate gene studies focus on specific genes that are selected based on a priori knowledge of their function, and compare the frequency of the variants between cohorts of individuals with and without disease.59 To date, a number of candidate-gene association studies have identified common variants that are more prevalent in AF populations compared with control populations.40,46–58 Often, the candidate genes have been selected based on results of studies in familial AF. As summarised in Table 2, a range of common variants in ion channel and non-ion channel genes have been associated with AF. Overall, however, candidate-gene association studies have been associated with limited success due to a low pre-test probability of the selected variants being involved in disease pathogenesis and poor reproducibility.59
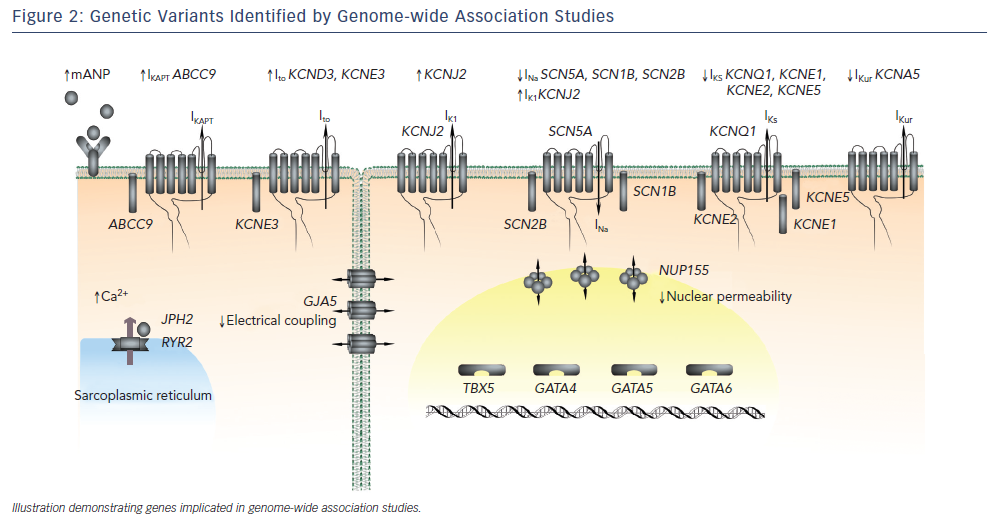
The recent advent of GWAS has led to significant progress in our understanding of the genetic basis of AF. GWAS involve genotyping up to a million common variants, or single nucleotide polymorphisms (SNPs), distributed throughout the genome and comparing their frequency between AF and control cohorts.60 As opposed to candidate gene studies, GWAS are unbiased and therefore may identify previously unsuspected genes that play an important role in disease pathogenesis.
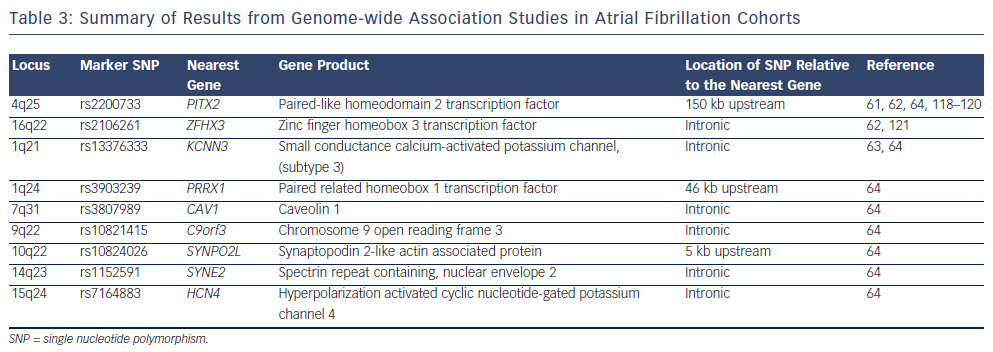
Four large GWAS have been performed in AF cohorts to date.61–64 The results of these studies are summarised in Figure 2 and Table 3. The common variants identified by GWAS are located either within or in proximity to compelling candidate genes for AF. The ion channel genes, KCNN3 and HCN4, have been identified at two of the GWAS loci for AF.63,64KCNN3 encodes a calcium-activated potassium channel (SK3 channel), which is abundantly expressed in the atrium.65HCN4 encodes the hyperpolarisation-activated, cyclic nucleotide-gated cation channel 4, which underlies the pacemaker potential.66 Two of the GWAS loci for AF harbour the cardiac transcription factor genes PITX2 and PRRX1. These homeobox transcription factors are critical mediators of cardiac development. PITX2 mediates asymetrical development of the heart and inhibits left-sided pacemaker specification.67–69PRRX1 has been implicated as a mediator of development of the pulmonary veins.70SYNPO2L, MYOZ1 and CAV1 also represent potentially interesting genes at GWAS loci. SYNPO2L and MYOZ1 encode signalling proteins that localise to the Z-disc and modulate cardiac sarcomeric function.71,72CAV1 is an important membrane protein, which plays a role in cellular signalling, and has been demonstrated to interact with ion channels, including HCN4 and KCNN3.73–76
In addition to the variants identified in the aforementioned GWAS for AF, common variants that have previously been implicated in GWAS for Brugada syndrome have also been demonstrated to influence risk in AF. AF is commonly observed as a co-existing condition in pedigrees with Brugada syndrome.77,78 Paradoxically, variants that have been demonstrated to confer increased risk of Brugada syndrome have a protective effect in AF patients.79 Further functional studies are necessary to determine the mechanisms underlying these observations.
While compelling candidate genes have been identified at GWAS loci, it is important to note that the mechanistic link between the GWAS variants and the function of these genes represents a challenge. This point is underscored by the fact that, to date, more than 1,000 GWAS risk variants have been identified for a range of diseases, while only a handful have been comprehensively functionally validated.80 GWAS rarely identify causative genetic variants directly. Rather, the variants identified by GWAS typically act as markers that point to a disease causing variant.60 The prevailing theory regarding the mechanistic link between the causative variants at GWAS loci and disease pathogenesis is that these variants alter the quantity of target gene expression, possibly through altered function of transcription regulatory elements.81
Interestingly, researchers have recently demonstrated that the rare variants identified in population-based genetic studies may contribute to variable penetrance of causal mutations in familial forms of AF. In a study involving 11 AF pedigrees, in whom the causative mutation was known, Ritchie et al. demonstrated that the presence of the risk variants at the 4q25 locus predicted whether mutation carriers developed AF.82 As discussed above, variants at the 4q25/ PITX2 locus have consistently been demonstrated to influence AF in multiple population-based studies. These findings suggest that the heritability of AF is influenced by complex interactions between common and rare variants.
Clinical Relevance
The identification of genetic variants that contribute to AF susceptibility has potentially important implications for management of the arrhythmia. On the one hand, these variants could be of value for determining risk of future AF in asymptomatic individuals. On the other, they could uncover novel molecular targets for pharmacotherapy and potentially be of use in predicting response to therapy in AF patients. The following section discusses the potential utility of genetic information for management of AF patients.
Risk Stratification for Atrial Fibrillation The identification of asymptomatic individuals who are at high risk of developing AF is an important public health concern. Current risk-stratification strategies, which are based mainly on conventional risk factors, are associated with significant limitations.83–86 Following the success of GWAS, the potential use of genotype-based risk stratification for AF has received significant interest. Initial attempts at using GWAS risk variants to predict risk have been associated with limited success. For instance, Smith et al. demonstrated that when considered in combination with conventional risk factors, genotype information from two AF GWAS loci (chromosome 4 and 16) did not have an impact on risk stratification.87 Similarly, Everett et al. reported that the inclusion of 12 risk variants at nine GWAS loci did not significantly enhance risk stratification.88 However, more recently, encouraging results have emerged from a study by Lubitz et al. who demonstrated that when considered in combination, four risk variants at the 4q25/ PITX2 locus and eight variants at other loci resulted in a fivefold gradient in AF risk.89 The results were consistent in cohorts of European and Japanese descent.
It is important to note that the variants identified to date are associated with modest effect sizes and collectively account for only a proportion of the heritability estimated for AF. Before clinically applicable risk stratification algorithms can be developed for AF, a significant proportion of the ‘missing heritability‘ of AF needs to be uncovered. The identification of the missing heritability of complex phenotypes like AF represents a major hurdle. Some of the missing heritability may be accounted for by additional common variants. GWAS are designed to identify common variants; however, some common variants may have been overlooked as current thresholds for statistical significance are high. Potential strategies for the identification of additional common variants include performing GWAS in larger cohorts and cohorts from different ethnic backgrounds.90
A significant proportion of the missing heritability of AF is likely to be accounted for by rare variants and structural variants in the genome.90 Examples of structural variation in the genome include tandem repeat sequences, insertions and deletions, copy number variants, translocations and inversions. GWAS are not designed to identify rare variants or structural variants. As discussed previously, a number of novel genotyping technologies have emerged since GWAS, including exome sequencing and whole genome sequencing. These represent promising techniques for the identification of rare variants underlying complex traits at a population level. While genotyping techniques for the identification of structural variants are currently less effective, they are constantly evolving.
Identification of Novel Therapeutic Targets for Atrial Fibrillation
The identification of the genetic architecture underlying AF has the potential to uncover novel therapeutic targets for the arrhythmia. GWAS are of particular interest in this context as they are agnostic and therefore commonly identify previously unsuspected genes underlying complex traits. As discussed previously, GWAS have identified a number of compelling candidate genes at the AF risk loci. These findings have spawned additional functional studies that have focused on candidate genes and have demonstrated that genes such as KCNN3 and PITX2 influence AF susceptibility.91–96
It is important to note that while the aforementioned studies suggest that candidate genes at GWAS loci potentially influence AF susceptibility, focusing drug development efforts on these candidate genes would be premature. Before GWAS findings can be translated to drug development, more comprehensive functional validation is necessary. GWAS typically characterise the association between marker variants and AF. The aims of post-GWAS analysis include identification of the causative variants at the GWAS loci and characterisation of the mechanistic link between these variants and target genes. A detailed discussion of post-GWAS functional analysis is beyond the scope of this review and has previously been reviewed extensively.81
In addition to GWAS, exome sequencing and whole genome sequencing could potentially identify important therapeutic targets for AF. Population-based exome sequencing projects are already currently under way in AF cohorts and promise to identify multiple additional candidate genes. As is the case with GWAS however, these genes will have to be comprehensively functionally validated. Overall, while findings from population-based genetic studies are promising, it may take more than a decade of research from the discovery of a novel gene to the development of a drug that can be used in clinical practice.97
Genotype-based Prediction of Response to Atrial Fibrillation Therapies
In addition to uncovering novel therapeutic targets for AF, information from genetic studies could potentially be of value in predicting responses to established therapies. The influence of genotype on response to antiarrhythmic drugs has been investigated in two recent studies. In a relatively small cohort of AF patients, Parvez et al. demonstrated that the GWAS risk variants at the 4q25 locus independently predict successful rhythm control with antiarrhythmic therapy.98 The same group also demonstrated that variants in the gene encoding the β1-adrenergic receptor (β1-AR) predict response to rate control therapy with beta blockers.99
Multiple studies have reported that genotype also influences response to anticoagulant therapy. Variants in genes such as VKORC1, CYP2C9 and CYP4F2 have been identified as potential mediators of response to warfarin therapy.100,101 These genes encode proteins that are either involved in the vitamin K pathway or in warfarin metabolism.102 Attempts have been made to incorporate these genes into algorithms designed to predict warfarin response. While some studies have suggested potential clinical utility of these algorithms, the results have not been consistent.102 A more detailed discussion on pharmacogenomics of warfarin therapy is beyond the scope of this review.
The role of genotype-based prediction of therapeutic response has also been investigated for non-pharmacological interventions. Husser et al. demonstrated that the variants at the 4q25 locus predict response to catheter ablation for AF. Specifically the presence of the risk variants predicted both early and late recurrences of AF following pulmonary vein isolation.103 Further evidence linking SNPs at the 4q25 locus and response to catheter ablation came from a more recent study from Shoemaker et al.104 Finally, Parvez et al. reported that SNPs at 4q25 predict recurrence of AF following successful direct current cardioversion.105
Overall, the above studies have demonstrated promising results suggesting that genotypic data can be of value in to predicting response to both pharmacological and non-pharmacological therapies. However, most of these studies have been limited to small numbers of patients and have focused on small numbers of variants. Further research with large, prospective randomised studies that include multiple genetic variants is currently needed to more clearly define the role of genetic data in predicting response.
Conclusions
In recent years, research into the genetic basis of AF has undergone a revolution. Significant progress has been made in identifying the genetic substrate underlying the common form of AF encountered in everyday clinical practice. Genotyping technologies are constantly evolving and promise to uncover more genes and molecular pathways underlying AF. However, while these discoveries are promising, much work remains before they can be translated to the clinic.