







Short QT syndrome (SQTS) is a rare, inheritable channelopathy of the heart characterised by abnormally short QT intervals on the electrocardiogram (ECG) and an increased propensity to develop atrial and ventricular tachyarrhythmias in the absence of structural heart disease.1,2 SQTS was first described as a new clinical entity by Gussak et al. in 2000.1 Until then shortening of the QT interval had only been reported in the context of electrolyte imbalances (hyperkalaemia, hypercalcaemia), hyperthermia, acidosis and endocrine disorders. The familial nature and arrhythmogenic potential of the disease were further confirmed by Gaita et al.2 They described six patients with SQTS in two unrelated European families with a family history of sudden death in association with short QT intervals on the ECG. Since its recognition in 2000, significant progress has been made in defining the clinical, molecular and genetic basis of SQTS as well as the therapy options. Today, SQTS is usually defined as QTc ≤330 ms, or QTc interval <360 ms and one or more of the following: history of cardiac arrest or syncope, family history of sudden cardiac death (SCD) at age 40 or younger or a family history of SQTS.3 The purpose of this review is to summarise the available data and to discuss recent advances in the diagnosis and therapy of SQTS.
Establishing the Diagnosis – Electrocardiographic Criteria
The ECG constitutes the mainstay of the diagnosis of SQTS. The hallmark ECG finding is an abnormally short QTc. Although it may appear reasonable to assume that a shorter QTc could predispose to a higher risk for ventricular arrhythmias, to date, there is no evidence to support this hypothesis.4–6 Controversy exists about the exact cut-off value for the short QTc interval.
Population-based and genetic studies show that QTc interval <330 ms is extremely rare.6–8 Data from over 10,000 adults suggest that, in the healthy population, the prevalence of QTc <340 ms is approximately 0.5 % (with 95 % confidence interval).6 Therefore, males with QTc ≤330 ms and females with QTc ≤340 ms have abnormally short QT and should be considered to have SQTS, even if they are asymptomatic. However, it should be emphasised that the prognosis of patients with asymptomatic SQTS still remains undefined. Individuals with QTc <320 ms who reached adulthood without developing life-threatening arrhythmias have been reported.7,9 Anttonen et al.6 further reported a low rate of all-cause mortality in individuals with QTc intervals <320 ms. However, since only middle-aged subjects were included in their study, the findings may not be applicable to a younger population. On the contrary, subjects with a ratecorrected QT interval >450 ms had greater all-cause and cardiovascular mortality than those with normal or short QT intervals. Nielsen et al.10 recently reported an increased risk for cardiovascular death in patients with QTc intervals <379 ms. The hazard ratio was more pronounced in women than in men and in patients of 50–70 years of age.
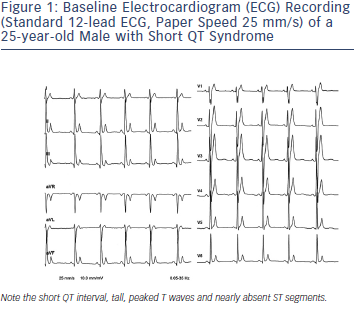
Population studies also show that relatively small number of individuals have QTc intervals <360 ms (males) and <370 ms (females), respectively, so that these values probably should be regarded as ‘short’. A diagnosis of SQTS should be considered when such patients present with cardiac arrest, unexplained syncope or atrial fibrillation (AF) at a young age. As for patients with QT prolongation, the first step in clarifying the diagnosis among patients with ‘short QT’ is performing repeat ECGs to further study the QT duration and T-wave morphology at different heart rates.11 When the diagnosis of SQTS is suspected, resting 12-lead ECG should be performed at a heart rate within normal limits. The QT interval should be measured when the heart rate is <100 bpm and preferably less than 80 bpm, because all QTc formulae will overcorrect the true QTc intervals at higher heart rates, leading to a false negative diagnosis. Holter monitoring or long-term ECG monitoring becomes necessary in such cases to make the correct diagnosis. In addition to the QTc interval there are several other ECG findings that may facilitate the correct diagnosis (see Figure 1):
- The QRS complex is directly followed by a T wave; ST segment is usually absent.12
- T waves are tall, peaked, symmetrical and narrow-based
- Rate-dependent prolongation of the QT interval at slow heart rates is abrogated in SQTS patients, remaining below the lower limit of normal values.13
- Often a prominent U wave can be observed, separated by an isoelectric T–U segment.14
- Longer Tpeak – Tend interval may be observed, suggestive of augmented transmural dispersion of refractoriness.15
- Depression of the PQ segment, due to a heterogeneous abbreviation of atrial repolarisation, most prominently in inferior and anterior leads.16
Molecular Mechanism/Genetics
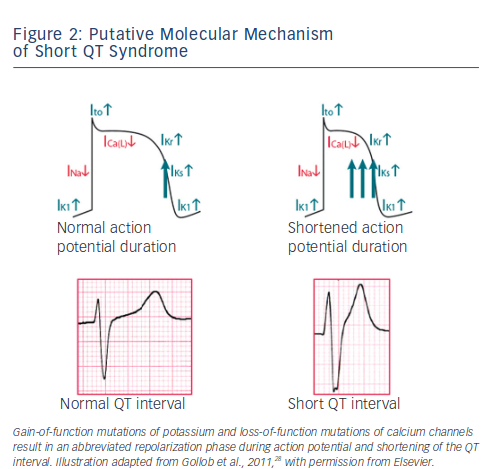
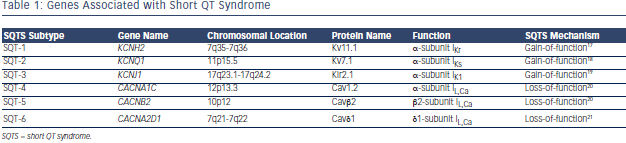
SQTS is a rare, sporadic or autosomal dominant disorder characterised by markedly accelerated cardiac repolarisation and manifested by atrial and ventricular arrhythmias, and/or SCD. To date, causative mutations in potassium17–19 and calcium channel genes20,21 have been identified. Gain-of-function mutations of potassium and lossof- function mutations of calcium channels result in an abbreviated repolarisation phase during action potential and shortening of the QT interval (see Figure 2). Because of heterogeneous contribution of repolarising ion currents within the heart, there is an inhomogeneity and dispersion of repolarisation, which provides substrate for the development of both atrial and ventricular tachyarrhythmias. Three main genetic variants have been described in the SQTS (see Table 1), involving potassium channel genes also associated with the long QT syndrome (LQTS). However, while mutations in the potassium channel genes causing LQTS are loss-of-function mutations, those observed in SQTS are gain-of-function mutations.
The SQTS subtype SQT-1 is caused by mutations in KCNH2 (HERG), the gene also responsible for LQT-2. Genetic screening of the first two reported families with SQTS and SCD led to the identification of two different missense mutations in KCNH2 that caused the same amino acid change in the cardiac Ikr channel.17 In one family, a missense mutation with a cytosine to guanine substitution at nucleotide 1764 on KCNH2 was reported, while in the second family a cytosine to adenine substitution at the same nucleotide was observed. Both mutations led to a substitution of asparagine at codon 588 with a positively charged lysine.
To further elucidate the mechanism of QT interval shortening, the mutated KCNH2 channel (N588K) was co-expressed with and without the ancillary β-subunit MiRP1 (KCNE2) in human embryonic kidney cells (TSA201) and patch-clamp experiments were performed. Whole-cell recordings demonstrated that the N558K missense mutation abolished rectification of the current at plateau voltages, which results in a significant increase of Ikr during the early phases of the action potential and leads to an abbreviation of the action potential and thus to abbreviation of the QT interval. Shortening of ventricular action potential is supposed to be linked to a shortening of the effective refractory period, thus causing an increased ventricular and atrial susceptibility to premature stimulation, leading to AF and ventricular fibrillation (VF).
Genetic heterogeneity in the SQTS was indicated by the findings of Bellocq et al.18, who identified a mutation in KCNQ1 (V307L) in a 70-year-old patient with abnormally short QTc duration (302 ms) and aborted SCD. Similar to the findings by Brugada et al.,17 the mutation in KCNQ1 caused a gain of function of IKs, resulting in an abbreviation of the action potential duration and shortening of the QT interval in vitro. The KCNQ1 gene is therefore not only responsible for LQT-1, but also for SQT-2. A missense mutation in KCNQ1 was reported in a neonate presenting with in utero bradycardia and postpartum short QT intervals and AF.22 To characterise the physiological consequences of the V141M mutation, Xenopus oocytes were injected with complimentary RNA (cRNA) encoding wild-type KCNQ1 or mutant V141M KCNQ1 subunits, with or without KCNE1. Computer modeling showed that the mutation would accelerate the activation kinetics of human ventricular myocytes and abolish pacemaker activity of the sinoatrial node, by a loss of voltage-dependent channel gating.22
In 2005, SQT-3 was introduced by Priori et al.19 and was associated with a novel gain-of-function mutation in the KCNJ2 gene, encoding for the strong inwardly rectifying channel protein Kir2.1. The loss-of-function mutations of KCNJ2 are responsible for LQT-7. In two affected family members a single base-pair substitution (G514A) in the KCNJ2 gene could be identified, resulting in an amino acid change from aspartic acid to asparagine at position 172 (D172N) in the Kir2.1 potassium channel. Functional characterization of the mutation demonstrated a significant increase in the outward IK1 current, and strongly suggests that myocardial tissues carrying the D172N mutation in the Kir2.1 channel should be capable of sustaining stable functional reentry at high frequencies.19
Finally, Antzelevitch et al.20 reported three cases in which the Brugada Syndrome phenotype and a family history of SCD was combined with shorter-than-normal QT intervals (≤360 ms). In these three cases a mutation in genes encoding the α1- or β2b- subunits of the cardiac L-type calcium channel were identified, with three specific mutations in CACNB2b (S481L) and CACNA1C (A39V and G490R) declared responsible for the ECG phenotype. To determine the contribution of each mutation to the clinical phenotype, each of the wild-type and mutated CACNA1C and CACNB2b mutations were expressed in Chinese hamster ovary cells. The results of patch-clamp experiments indicate that all the mutations cause a major loss of function in calcium channel activity.20 The QTc observed in these three cases and in affected family members ranged from 330 to 370 ms, a longer QTc interval than that observed in SQTS subtypes 1–3. Another overlapping phenotype of short QT interval and Brugada-like ECG has been reported in a 40-year-old Chinese patient with family history of SCD.23 The QT interval was 320 ms and the ECG showed a Brugada-like pattern in precordial leads.
Lastly, Templin and co-workers21 introduced SQT-6, when they reported a novel mutation in the CACNA2D1 gene, which presumably led to short QT interval (QTc 329 ms) and documented VF in an otherwise healthy 17-year-old female. Functional analysis showed that the discovered missense mutation (p.Ser755Thr) in CACNA2D1 decreases the current of the L-type calcium channel, which is responsible for the cardiac action potential plateau phase and the cytoplasmic Ca2+ transients regulating the contraction force.24 Whether mutations in CACNA2D1 may cause other manifestations of J-wave syndrome remains to be investigated.25 A systematic overview of causative genes in SQT1-6 is shown in Table 1.
{kind=link}
From Bench to Bedside – Clinical Presentation of SQTS
SQTS has been described only in few families worldwide. There have been approximately 80 SQTS cases reported so far. All probands presented with a QTc below 320 ms without evident structural heart disease. In the largest available case series of 45 genetically screened and followed SQTS patients reported recently, most patients have experienced symptoms.5 Often the first symptom of the disease may be lone AF in the absence of structural heart disease. However, ventricular arrhythmias and sudden cardiac arrest may also be the first symptom, which makes early diagnosis challenging.
The available data suggest that patients are at risk throughout their lifetime, with a peak between the second and fourth decades.26 This distribution is thought to be due to the peak in testosterone plasma levels, which may cause shortening of the QT interval, especially in male adolescents. Mean age at diagnosis is 23 years, with predominant occurrence in male patients. Cardiac arrest seems to be the most frequent presenting symptom (up to 40 %).5 Palpitations are very common (30 %), followed by syncope (25 %) and AF, which is the first presenting symptom in up to 20 % of patients. Forty-seven percent of patients were asymptomatic and were diagnosed due to strong family history. Strong family history of arrhythmic symptoms including SCD is a common finding. The circumstances of onset of symptoms are highly variable, and episodes of SCD have been reported during or following loud noise, at rest, during exercise and during daily activities.4 The yield of genetic screening is low and varies between 15 % and 25 %, despite the fact that a familial association is present in the majority of patients.
Risk Stratification in SQTS – Does QT Length Really Matter?
The greatest challenge for the management of patients with SQTS remains the paucity of risk identificators. Scoring systems that include clinical and ECG criteria are established in the case of congenital long QT syndrome.27 Correspondingly, the SQTS diagnostic criteria proposed by Gollob et al.28 are composed of four different components: ECG, clinical history, family history and genotype. In order to be eligible to receive points in the last three sections, a minimum of 1 point must be received from the ECG criteria. An overall score of 4 points or greater indicates a high-probability diagnosis of SQTS, whereas 2 points or fewer makes a diagnosis of SQTS a low probability. Patients with a score of 3 points are considered to have an intermediate probability of having SQTS.
In an effort to explore the risk-stratifying prognostic value of the Gollob score, a modification of the proposed diagnostic criteria was suggested to predict events in a paediatric cohort of SQTS.29 Twenty-five paediatric SQTS patients were followed over six years, during which time 56 % (14 patients) had symptoms (survived cardiac death, syncope and AF). Subjects with a modified Gollob score of ≤3 corresponded to a low probability of cardiac events, whereas patients with a higher score were more often symptomatic. In contrast to these findings, Mazzanti et al.5 recently reported a cohort of 47 SQTS patients and 26 affected family members, who were enrolled and followed over a mean period of five years. The observed annual event rate for cardiac arrest was 10.6 % among patients who already experienced cardiac arrest, and only 0.4 % in patients without history of cardiac arrest at initial presentation. The authors did not observe a relationship between the original28 and prognostic29 Gollob score and a probability of cardiac events, as the majority of subjects with a score ≤3 (low risk) experienced cardiac arrest and should therefore be regarded at high risk. However, history of survived cardiac arrest at the initial presentation proved to be a strong predictor of recurrent ventricular arrhythmias over the course of time. As emphasised by the authors, this information is important when considering the indication for implantable cardioverter defibrillator (ICD) therapy especially in young SQTS patients or infants with history of survived cardiac arrest.5 The high rate of cardiac events in the Mazzanti et al. cohort further corroborates observations from earlier studies4,26,29 and underscores the malignant nature of SQTS. However, one must also take into account a high probability of inappropriate ICD shocks in paediatric patients, which in the study of Villafane et al.29 by far exceeded the rate of appropriate shocks. Finally, Anttonen et al.30 observed repolarisation differences between symptomatic patients and asymptomatic individuals with shortened QT intervals. The authors concluded that an interval measured from the J point (Jp) to the highest point of the T wave (Tpeak) less than 150 ms identifies symptomatic SQTS patients from asymptomatic individuals with short QT intervals.
Therapeutic Options
As with other inherited channelopathies, there are principally two options for therapeutic interventions. Due to the malignant nature of SQTS, the ICD is recommended in symptomatic SQTS patients who are either survivors of sudden cardiac arrest and/or have documented spontaneous sustained ventricular tachyarrhythmias with or without syncope (Class I recommendation).3 A unique problem with ICDs in SQTS stems from one of the syndrome’s main features on ECG: the tall and peaked T wave that closely follows the R wave can sometimes be interpreted as a short R–R interval, provoking an inappropriate shock from the ICD.31
The high prevalence of AF in SQTS patients may present a clinical challenge for appropriate ICD programming. Reprogramming the decay delay, sensitivity or both generally prevents inappropriate discharges. Villafane et al.29 reported a remarkably high rate of inappropriate ICD therapies of up to 64 % in six years in a large paediatric cohort of SQTS patients, which accentuates the need for sophisticated and durable ICD devices.
Although the ICD remains the mainstay of therapy for SQTS, pharmacological therapy may be useful as an adjunct to the ICD or may be used for primary prevention in cases in which the patient refuses an ICD or in young children in whom the implantation of an ICD may be problematic.
Gaita et al.32 tested four different antiarrhythmic drugs including flecainide, sotalol, ibutilide and hydroquinidine in six patients with SQT-1. Only hydroquinidine prolonged the QT interval to normal levels, increased ventricular ERP and rendered VF noninducible. Class Ic and III antiarrhythmic drugs failed to do so. Quinidine also restored the QT–RR relationship toward the normal range.33 In a one-year followup, patients treated with hydroquinidine remained asymptomatic and no further episodes of ventricular arrhythmia were detected. Schimpf et al.34 reported clinical efficacy of disopyramide, another Class Ia antiarrhythmic drug, in two patients with SQT-1, consistent with the experimental data from heterologous expression of N588K KCNH2 mutant channels. Oral administration of disopyramide in these patients increased the QT interval and ventricular refractory period and abbreviated the Tpeak–Tend interval. Other class III antiarrhythmic drugs including d-sotalol, amiodarone and nifekalant were also sporadically reported to be effective in treating VT/VF storm and prolonging atrial and ventricular effective refractory periods and normalising the QT interval.35,36
AF is a common clinical problem in SQTS. Some SQTS patients exhibit only AF.37 Propafenone has been shown to be effective in preventing frequent paroxysms of AF with no recurrence of arrhythmia for more than two years without any effect on QT interval.38
In summary, the pharmacological therapy of patients with SQTS is still poorly defined, mainly due to a lack of large-scale evidence. Hydroquinidine seems to be the first-line therapy for SQTS patients and based on the guidelines3 should be considered the most effective pharmacological therapy in SQTS.