










The health benefits of exercise are well established and extend beyond the cardiovascular system.1 These benefits accrue from the modulation of traditional risk factors for atherosclerotic cardiovascular disease, as well as through an anti-inflammatory effect on the vascular endothelium and changes in autonomic regulation.2 A meta-analysis in almost 900,000 individuals demonstrated that the physically active group had a 35% reduction in the risk of cardiovascular death and 33% reduction in all-cause mortality.3
The WHO recommends a minimum of 150 minutes of moderate-intensity exercise or 75 minutes of vigorous-intensity exercise per week. 4 A cohort of nearly 650,000 individuals participating in physical activity at half these recommended levels, at the recommended levels and at three times the recommended levels, gained 1.8, 3.4 and 4.3 years of life, respectively.5 Higher cardiorespiratory fitness levels correlate with greater benefit, with a mortality risk reduction of 13% for each additional metabolic equivalent (MET) increase in exercise capacity.6,7 These data suggest that, at the population level, a greater volume of exercise results in greater cardiovascular benefit. A more cautious approach is necessary in individuals with established heart disease, where the volume and intensity of exercise may need to be moderated.8
Endurance athletes routinely exercise far beyond the WHO recommendations.9,10 The sustained elevation of cardiac pressure and volume loads associated with regular exercise promote a series of electrical, structural and functional adaptations, collectively termed ‘athlete’s heart’. The nature and magnitude of changes vary by sporting discipline, ethnicity, age and sex, and can overlap with mild phenotypes of conditions associated with arrhythmias and sudden cardiac death (SCD).11 Extreme cavity dilatation, left ventricular (LV) hypertrophy, elevated coronary artery calcium (CAC) scores, acute cardiac biomarker release, myocardial fibrosis and cardiac arrhythmias have all been reported, raising concern of a reverse U-shaped relationship between the volume of exercise and cardiovascular health, with diminishing cardiovascular benefit and potential harm.12–15 Therefore, there is ongoing debate as to whether there is a threshold that constitutes ‘excess of exercise’, which may induce harm. To separate myth from reality, this review reports on the evidence supporting the notion of ‘too much exercise’ and the proposed mechanisms of exercise-induced cardiac arrhythmias in ostensibly healthy athletes.
AF
AF is the most common sustained arrhythmia in the general population; it is a major cause of ischaemic stroke, heart failure and impairment in cognition and quality of life, and increases the risk of death.16–18 The incidence of AF increases with age, given that age in itself is a determinant of AF. Moreover, advanced age is associated with cardiovascular risk factors, heart failure, structural heart disease, coronary artery disease and chronic kidney disease, all of which are linked with an increased risk of AF.19 It is well established that exercise mitigates such risk factors and, as such, regular exercise can prevent AF onset, as well as also improve symptoms, morbidity and mortality in those with established AF.20–22 A study of 6,000 veterans with a mean age of 56.8 years undergoing a symptom-limited exercise tolerance test found exercise capacity to be inversely related to the incidence of AF during a median follow-up of 8 years. The fittest individuals were found to have the lowest risk of developing AF, with a 21% decrease for each MET increase in exercise capacity.20 The Cardiovascular Health Study of 5,446 adults aged >65 years identified greater leisure time activity and walking as being associated with a lower incidence of AF, with progressively lower risk with greater activity levels, and a 44% risk reduction in those undertaking moderate physical activity.23 Importantly, however, risk reduction diminished in those undertaking high-intensity exercise (>6 METs).23

An emerging body of evidence has since supported a link between long-term intense endurance exercise and AF in an ‘exercise paradox’ (Figure 1). Larger epidemiological studies and several meta-analyses have demonstrated that the incidence of AF is two- to fivefold greater in endurance athletes than in non-athletes.24–29 The elevated risk in the athletic group dissipates with increasing age (>55 years) and the presence of cardiovascular risk factors. There is evidence to support the notion that exercise intensity, duration and type of sport affect the onset of AF. In a study of 52,755 cross-country skiers participating in a 90 km cross-country skiing race, the participants who completed more than five races were at highest risk of AF, and were more likely to develop AF than those who undertook one race (HR 1.29).24 Similar findings have been observed among healthy, middle-aged male physicians, with those participating in higher-intensity jogging having a 53% higher risk of AF compared with men who did not exercise.30 This would suggest that the association between exercise and AF is not restricted to elite athletes, and is also observed in the general population. However, the exact dose of exercise that confers risk of AF remains unclear, with high-quality prospective studies with well-defined study populations still lacking. A figure of around 1,500–2,000 lifetime exercise hours has been suggested as the threshold at which AF risk increases, with a peak age of onset at >40 years.31 AF in younger athletes is unusual and should prompt evaluation for underlying heart disease.32,33
Most studies investigating the relationship between AF and exercise have focused on male elite athletes, who historically dominated the landscape of elite sports. The link between exercise and AF in female athletes is less clear. In a large cohort of more than 140,000 male and 160,000 female athletes, increasing levels of physical activity were associated with AF in male, but not female, participants.34 A meta-analysis of 22 studies identified an increased risk of AF in men undertaking intense exercise but, conversely, intense exercise was protective in women.25 Similarly, a more recent meta-analysis also concluded that the general risk of AF is lower in female than male athletes.27 However, there remains a lack of data on high-level female endurance athletes, who would surpass the level of exercise undertaken by the female participants of these studies.
The mechanism of AF in athletes is not well understood, with much of our knowledge based on animal models. Vagal tone, which is chronically elevated in athletes, is thought to be one of the most important contributors to the development of AF.35 In addition, atrial remodelling, in the form of atrial dilatation and fibrosis, is increasingly being recognised as an important factor. Atrial remodelling in athletes is considered to be a physiological response to exercise, because the overall reservoir function appears to be preserved with atrial dilatation; however, given that atrial dilatation in pathological conditions contributes to the development of AF, it remains to be seen how distinct atrial remodelling in athletes is from that seen in pathological states.36,37 AF episodes are most common during states of increased parasympathetic tone (rest, sleep), but sympathetic stimulation during exercise may also trigger AF, in association with atrial wall stretch and inflammatory cytokines.15 In a study of rat models of chronic endurance exercise, AF was induced after 16 weeks of training with identifiable atrial dilatation, fibrosis in the atria and right ventricle (RV) and autonomic changes, which did not fully resolve with detraining.14
Bradyarrhythmias
Sinus bradycardia and sinus pauses are common in endurance athletes. In a study of 62 former professional male cyclists, compared with 62 well-matched controls (male golfers), the former endurance athletes demonstrated more frequent sinus bradycardia, sinus node dysfunction and pacemaker implantation for bradyarrhythmias relative to the control group.38 This is widely believed to be a consequence of high vagal tone, although, because these findings can persist despite detraining, adverse remodelling and fibrosis of the conduction system are also thought to be contributing factors.38 More recently, evidence suggests significant electrical remodelling within the sinus node, with downregulation of potassium/sodium hyperpolarisation activated cyclic nucleotide-gated channel 4 (HCN-4).39,40 A possible dose–response relationship has also been suggested, with a study of cross-country skiers demonstrating that those who participated in more races had a higher risk of sinus node disease or third-degree atrioventricular block.24
Ventricular Arrythmias
Premature Ventricular Beats
Premature ventricular beats (PVB) are fairly common in athletes and are usually benign. However, they may be the only sign of heart disease, often leading to comprehensive evaluation. It is well established that PVBs may reflect the broader phenotype of cardiomyopathies and help differentiate pathology from physiological adaptation to exercise, particularly in athletes with mild phenotypic expression, often referred to as the ‘grey zone’.
Data supporting the notion that PVBs are more frequent in athletes and may represent a feature of athletic adaptation are contrasted by studies that show similar burden of ectopy in athletic and non-athletic individuals.41–43 Comparisons between studies are challenging due to differences in methodologies to record and report PVBs, as well as the absence of a standardised protocol guiding further investigation. PVBs have been reported in up to 14% of young athletes and 26% of veteran athletes, with no convincing association between sporting discipline, volume or intensity of exercise, years of sports participation and burden or complexity of PVBs.42–44 Furthermore, the overall burden of PVBs increases with age. These findings do not support the hypothesis that endurance sports activity increases the burden of ventricular arrhythmias.42–44
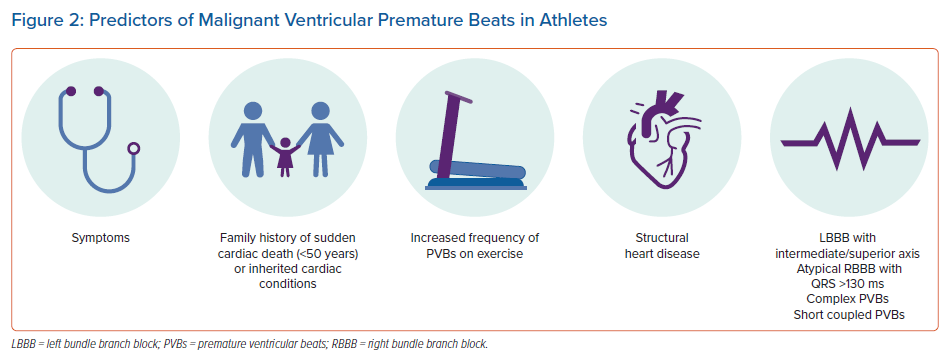
The PVB characteristics that imply association with disease are evolving (Figure 2). Traditionally, a frequency in excess of 2,000 PVBs/24 hours has been considered a red flag.41 Recently, however, evaluation of the morphology of PVBs, as a surrogate of ventricular origin, has emerged as the key factor in differentiating benign from potentially sinister PVBs.44–46 Frequent PVBs as a result of focal automaticity, emerging from the outflow tracts or from the fascicles of the left bundle branches, are relatively common and, in the absence of structural heart disease, should be considered benign.42,43 Other morphologies, such as PVBs with left or wide right bundle branch block or with intermediate or superior axis, are relatively uncommon and should be investigated further.46–48 Similarly, short coupling intervals, increasing PVB frequency during exercise and multifocal ectopy should prompt further evaluation. In particular, exercise-induced PVBs with multiple and/or alternating morphologies (bidirectional) may raise suspicion of underlying catecholaminergic polymorphic ventricular tachycardia.49
Effects on Ion Channels
Regular exercise exerts a significant effect on the expression and function of cardiac ion channels. Athletes exhibit longer QT intervals than sedentary individuals, with corrected QT intervals of 470 ms in male athletes and 480 ms in female athletes accepted as the upper limits of normal.8 Exercise-induced QT prolongation may confer an increased risk in individuals with underlying long QT syndrome (LQTS) because adrenergic surges and emotional stress may trigger arrhythmias in LQT1 and LQT2, respectively.50–53 Moreover, exercise-induced prolongation of the QT interval may pose considerable challenges in differentiating physiological adaptation from congenital LQTS, and potentially offering false reassurance to athletes at risk. A recent study demonstrated an exercise-induced QT prolongation phenotype, mimicking congenital LQTS, which reverts back to normal after a period of detraining.54 Although no arrhythmic events were recorded, more data are needed to fully understand the arrhythmic risk in individuals with acquired QT prolongation.54
Similarly, repolarisation patterns on the athlete’s ECG may overlap with the Brugada phenotype, causing a diagnostic conundrum.55 Although there are no clear data supporting a relationship between exercise and SCD in patients with Brugada syndrome, enhanced vagal tone at rest and in early recovery following exercise has been postulated as a precipitant of arrhythmia in athletes with Brugada syndrome.56
The Left Ventricle
Elevations in cardiac preload and afterload with chronic exercise are associated with cardiac chamber enlargement, with a 10–20% increase in wall thickness and 10–15% increase in ventricular cavity dimensions. Consequently, differentiation between athletic adaptation to exercise and a mild phenotype of primary cardiomyopathies may be challenging even for the most experienced of sports cardiologists. Male endurance athletes are typically observed with the largest cavity dimensions, with up to 14% exceeding 60 mm, a threshold that typically raises suspicion of a primary dilated cardiomyopathy.57 Ethnicity is important to consider in the evaluation of LV wall thickness. For example, an LV wall thickness of >13 mm is rare among white athletes, whereas it is more prevalent in black athletes (2% versus 12%, respectively).55,58 Crucially, regardless of ethnicity, a maximum wall thickness exceeding 16 mm is uncommon and should prompt consideration and further evaluation for hypertrophic cardiomyopathy. In addition, LV cavity dilatation and hypertrophy may persist in up to 20% of athletes, despite detraining, suggesting that extremes of cardiovascular adaptation to exercise may be irreversible.59 In a study by Finocchiaro et al., none of the first-degree relatives of decedents with unexplained LV hypertrophy (30% competitive athletes) were diagnosed with hypertrophic cardiomyopathy, suggesting that extreme LV hypertrophy may be a source of arrhythmias.60
The Right Ventricle
At rest, the RV functions against a very low resistance and high compliance pulmonary circulation. However, during exercise, RV wall stress increases 30-fold, reflecting a minimal reduction in pulmonary vascular resistance and a significant rise in pulmonary artery systolic pressures. This raises the possibility that repetitive intense exercise can induce structural changes and arrhythmias overlapping with arrhythmogenic right ventricular cardiomyopathy (ARVC), referred to as ‘exercise-induced ARVC’.61
Data from an animal model of endurance training demonstrated training-dependent RV fibrosis and tendency to arrhythmia following a 16-week exercise regime, which reversed after 8 weeks of exercise cessation.62 In a study of more than 300 athletes, RV enlargement meeting criteria for ARVC was seen in up to 45% of black athletes and 59% of white athletes, although none was diagnosed with ARVC.63 Studies have also reported transient RV dysfunction following endurance exercise, with greater dysfunction associated with more prolonged intense exercise, such as ultra-endurance events. In most studies there was no associated LV dysfunction, but there was correlation between the degree of RV dysfunction and elevation of troponin levels.64–66 Moreover, an evaluation of 46 endurance athletes presenting with arrhythmias by Heidbüchel et al. reported that 80% of arrhythmias were of RV origin and 89% of athletes fulfilled either definite (59%) or borderline/possible (30%) diagnostic criteria for ARVC.67 During a median follow-up of 5 years, 40% of athletes experienced major arrhythmic events defined as SCD, ICD shock or ventricular tachycardia. Subsequent genetic analysis of genes associated with ARVC, identified pathogenic variants in only 12.8% of athletes, compared with 30–50% expected in ARVC.68 Although these studies support the notion of exercise-induced ARVC, it is important to note that they included a highly selected cohort of athletes presenting with ventricular arrhythmias, and the genetic yield in ARVC may be far lower than 50% in the context of sporadic rather than familial disease. Moreover, other studies in elite Olympic athletes competing over many years have failed to demonstrate significant pathological RV remodelling, suggesting that this may be applicable to the very extremes of endurance training in individuals with some genetic predisposition, although it may not represent the classic ARVC genotype.69
By the same token, repetitive exercise in those with an established diagnosis of ARVC is well recognised to increase the risk of SCD through the acceleration of RV dysfunction and induction of ventricular arrhythmias.70,71 A North American multidisciplinary study reported that patients engaging in competitive sports were at a twofold increased risk of ventricular tachyarrhythmias or death and earlier presentation of symptoms than patients who participated in recreational sports and sedentary individuals.72 Similar results have been confirmed in desmosomal mutation carriers with no phenotypic expression, underscoring the impact of exercise on the RV.73 Further studies and longitudinal data are required to better understand the interplay between exercise and the RV in health and disease states.
Myocardial Fibrosis
In patient populations, the presence of late gadolinium enhancement (LGE) is an established adverse risk factor for malignant arrhythmia, and in athletes has been associated with a risk of complex VA.47,48,74,75
A small number of studies have demonstrated myocardial fibrosis in ostensibly fit male masters athletes engaging in endurance exercise. In a study of 102 middle-aged marathon runners, 12% demonstrated myocardial fibrosis (compared with 4% of controls), of which 42% demonstrated a pattern consistent with MI predominantly in the territory of the left anterior descending artery.15 Furthermore, there was suggestion of a dose–response relationship because participation in a greater number of marathons was an independent predictor for the presence of LGE.76 Similarly, in a study of 106 male masters endurance athletes, 14% demonstrated myocardial fibrosis, with almost half demonstrating a pattern consistent with a previous MI.13 Of those with evidence of MI, only half demonstrated coronary stenosis in the relevant coronary artery, raising the possibility of subclinical infarction, due to demand ischaemia, coronary spasm or plaque rupture.13
In a study of 83 asymptomatic middle-aged triathletes, participation in longer swimming distances and cycling races was an independent predictor for the presence of non-ischaemic LGE, affecting 17% of male athletes but none of the female athletes.77 A recent meta-analysis concluded that the incidence of LGE was almost sevenfold higher in middle-aged endurance athletes compared with non-athletes, with most of this due to mid-myocardial or subepicardial LGE, with the next most common pattern being insertion point fibrosis.78 Further longitudinal studies are required to better understand the temporal association of non-ischaemic fibrosis with acquired risk factors, such as an episode of myocarditis, and its clinical relevance in masters athletes. This is relevant in the era of the COVID-19 pandemic, which has ignited interest about the prevalence and potential implications of asymptomatic (subclinical) myocardial inflammation in elite athletes. A recent registry of 1,597 competitive collegiate athletes infected with COVID-19 reported symptomatic (clinical) myocarditis in five athletes (0.3%).79 The routine use of cardiac MRI (CMR) in all athletes increased the diagnostic yield of myocarditis by 7.4-fold to 2.3%.79 Importantly, follow-up CMR in 27 of the 37 athletes diagnosed with myocarditis (73.0%) demonstrated resolution of myocardial oedema (T2 elevation) in all, and LGE indicative of myocardial fibrosis in 11 (41%).79 Similarly, in a cohort of more than 3,000 athletes with COVID-19 infection, myocarditis was identified in 0.5% of those who underwent clinically indicated CMR following clinical assessment, but in 3% of the cohort of 198 athletes who underwent screening CMR.80
Coronary Artery Disease
Exercise is well established to reduce traditional risk factors for coronary artery disease, although masters athletes have been demonstrated to show elevated CAC scores, which is a powerful adjunctive predictor of future cardiovascular events in non-athletes.13,76,81 In a study of 152 masters endurance athletes with low Framingham risk scores (mean age 54 years), 19% of male athletes had a CAC score ≥100 Agatston units, compared with 4% among the controls, and 11% of athletes had a CAC score >300 Agatston units, compared with none among the controls.13 Furthermore, male athletes demonstrated twice as many atherosclerotic plaques (44% versus 22%), and 7.5% of male athletes demonstrated a luminal stenosis >50%, compared with none of the controls.13 Importantly, the significance of the elevated CAC scores may be mitigated by the plaque composition among athletes, which demonstrate a greater proportion of calcified plaques, which are considered more stable and less prone to rupture. In a study of 284 athletes, divided by lifelong exercise volume (<1,000, 1,000–2,000 and >2,000 MET-min/week), Aengevaeren et al. demonstrated that the most active athletes had a higher CAC score and more atherosclerotic plaque, but also a higher prevalence of calcified plaque.82 The longer-term longitudinal outcomes of endurance athletes remain unknown and further studies are warranted. In the Cooper Centre Longitudinal Study of more than 20,000 male participants, those performing >3,000 MET-min of exercise per week were more likely to have CAC, without increased all-cause or cardiovascular mortality after a decade of follow-up.83 Another study reported on 8,425 men who underwent an assessment of cardiorespiratory fitness and CAC and, over a 8.4-year follow-up, identified that each additional MET of fitness corresponded to a 14% lower risk of cardiovascular death in an adjusted model and attenuated the risk associated with higher CAC levels.84
Conclusion
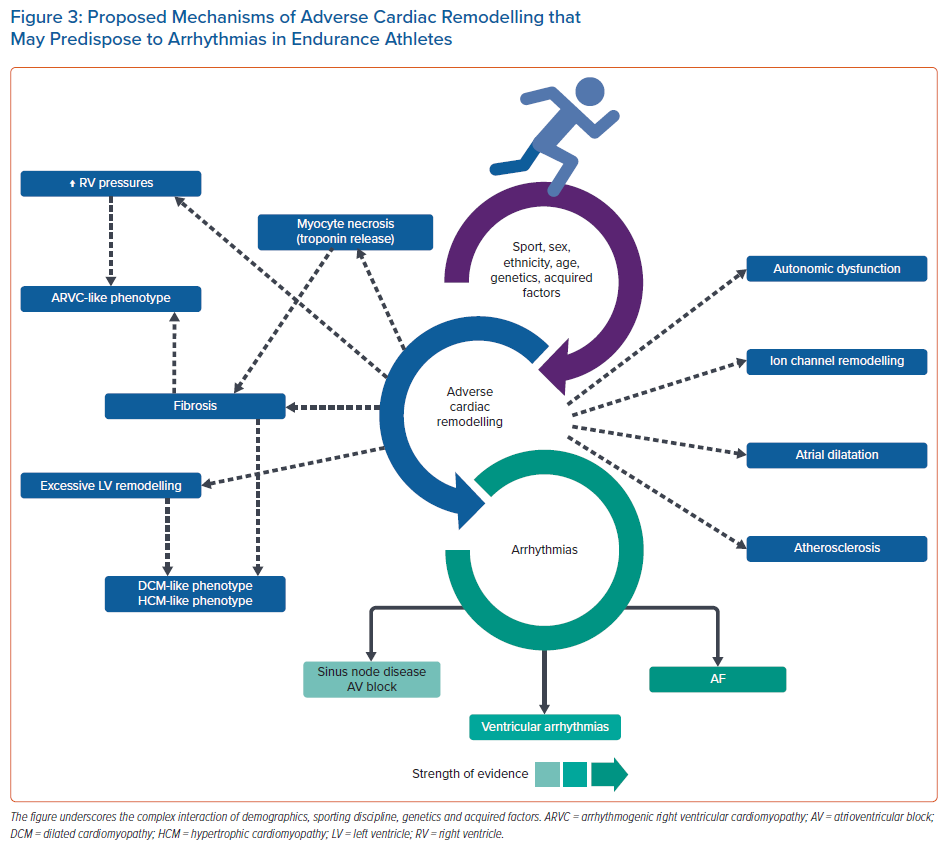
Exercise remains one of the most potent, cost-effective treatments against cardiovascular disease and cardiovascular mortality. Currently, evidence suggests that even high-intensity, high-volume exercise, and the associated lifestyle of elite endurance athletes, confers significant benefits, with athletes gaining an average of 5–7 years of life compared with sedentary individuals.85 Life-threatening arrhythmias remain overwhelmingly low, and mostly reflect underlying hereditary or congenital cardiac disease. Nevertheless, extremes of exercise may pose detrimental effects in an ‘exercise paradox’, with several routes of enquiry that require further study (Figure 3). Life-long endurance athletes seem to be at increased risk of AF in their 40s and a small number who participate in the most extreme of endurance sports may be predisposed to RV-related arrhythmias. More research is needed in better-defined cohorts with long-term follow-up.
Clinical Perspective
- The incidence of life-threatening arrhythmias in endurance athletes is low, and commonly reflects hereditary or congenital cardiac disease.
- Extremes of exercise may pose a detrimental effect; the proposed mechanisms are complex, with several routes of further enquiry ongoing.
- Athletes are at a higher risk of developing AF than non-athletes, particularly in their 40s, with both mixed and endurance sports conferring risk.
- Premature ventricular beats are common in athletes and are usually benign. Although ventricular arrhythmias have been associated with an exercise-induced arrhythmogenic phenotype, this seems to be applicable to the very extremes of endurance training in individuals with genetic predisposition.
- Further research is needed to ascertain the long-term significance of autonomic regulation and ion channel expression in endurance athletes, including extreme structural adaptations, coronary calcification, myocardial fibrosis and acute biomarker release.