










Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a disease characterised by progressive replacement of myocytes with fibrofatty tissue. These changes create a substrate prone to ventricular arrhythmia (VA) and increased risk for sudden cardiac death (SCD). Although initially thought to affect only the right ventricle, it has since been well-recognised that left ventricular (LV) involvement is common, and sometimes predominant.1–3
There have been significant advancements in the recognition and identification of predictive risk factors of the major outcomes of ARVC: VA and SCD. These include patient-controlled risk factors, such as exercise restriction, and unmodifiable risk factors, such as mutation status. Risk factor identification is important because it lays the foundation for stratifying an individual patient’s risk. A patient’s specific risk stratification should be used to make an informed decision regarding ICD placement, which is one of the main cornerstones of ARVC treatment. However, the decision to place an ICD must balance the potential short- and long-term complications of ICD placement with the risk for VA and SCD in the individual patient. This article will provide an overview of ARVC and examine the factors that contribute to an individual patient’s risk for VA and SCD.
Overview of Arrhythmogenic Right Ventricular Cardiomyopathy
Epidemiology
The prevalence of ARVC ranges anywhere from 1 per 1,000 to 1 per 2,000, with a higher prevalence in specific regions of Italy (Padua, Venice). The mean age of first presentation in one large cohort study was 36 ± 14 years.4 The most common presentations included VA in 50% of patients and cardiac arrest in 11%. The median age at cardiac arrest was 25 years old. It remains an important cause of SCD in young patients, particularly athletes.3 These factors highlight the importance of early recognition and appropriate therapy in ARVC.
Many individuals diagnosed with ARVC have a family history of the disease, and it is typically transmitted through an autosomal dominant pattern.3,5 In most cases, ARVC is inherited with an autosomal dominant pattern with variable expression. Most mutations that are associated with ARVC code for desmosomal proteins. A pathogenic mutation can be found in approximately two-thirds of patients. The clinical manifestations of ARVC appear to be worse in men compared with women, and this is further discussed later in this article.
Diagnosis and Management
The diagnosis of ARVC is based on the 2010 Task Force Criteria.6 These diagnostic criteria consist of major and minor diagnostic criteria pertaining to characteristics of RV dysfunction, histopathology on endomyocardial biopsy, repolarisation and depolarisation abnormalities on ECG, history of arrhythmia in the individual, and family history of ARVC or SCD. Each category has major (2 points) and minor (1 point) criteria. A score of 4 is considered definite ARVC; 3 points is borderline ARVC; 1–2 points is possible ARVC and 0 points is not ARVC.
Accurate diagnosis based on the 2010 Task Force Criteria is the first step in ARVC management. Once the diagnosis is secured, the second step of management is determination of an individual’s risk for VA and SCD. This will help to facilitate decisions regarding ICD placement, and is a large focus of this article. The other three components of this five-step approach to ARVC management are the minimisation of ICD therapy; prevention of disease progression; and cascade screening of family members.

Pharmacotherapy, catheter ablation, and exercise restriction are used to address the third step of ARVC management, the minimisation of ICD therapy. Pharmacotherapies include β-blockers and anti-arrhythmic drugs. β-blockers are thought to be beneficial in almost all patients with ARVC. Patients with ARVC are particularly sensitive to catecholaminergic effects.7 Beta-blockers not only prevent VAs, but are also a cornerstone of management in patients who have heart failure (Class of Recommendation [COR] I). In addition to β-blockers, sotalol (COR IIb) is the most commonly used anti-arrhythmic agent, followed by flecainide (COR IIb) and amiodarone (COR IIb).8–10
When anti-arrhythmic drugs fail or are not tolerated, catheter ablation becomes an important treatment option. Catheter ablation has been shown to reduce ventricular tachycardia (VT) events but does not reduce SCD risk or improve survival. Notably, a recent large study with more than 400 patients demonstrated continuing high rates of recurrence at 1 year (59%; 95% CI [44–71%]) and at 5 years (74%; 95% CI [59–84%]) despite ablation, but that overall burden of VA was reduced.11 The origin of VT is most commonly epicardial, and epicardial ablation paired with or without endocardial ablation has been shown to be safe and effective in further reducing VT events.12,13 In addition to pharmacological therapy and catheter ablation, exercise restriction is critical. We have recently reported that the tertile of patients with ARVC who reduce their exercise to the greatest degree have a 90% lower risk of developing VA (HR 0.10; 95% CI [0.02–0.43]).14
The fourth component of ARVC management is the prevention of progression and development of heart failure. It has been well-established that ARVC is a progressive disease and that heart failure develops over time in more than 40% of patients.15 The risk of progression is addressed with pharmacological therapy (β-blockers and renin–angiotensin–aldosterone system [RAAS] blockade) and exercise restriction. Management of overt heart failure symptoms is similar to any aetiology of heart failure. Diuretics should be used for congestive symptoms, and close attention should be placed on electrolyte balances given this population’s high risk for arrhythmia. Guideline-directed medical therapy for heart failure with reduced ejection fraction including β-blockers and RAAS blockade should be initiated as appropriate.16 Cardiac transplantation is required in a significant subset of patients.17 Cardiac transplantation is generally needed more than 15 years after initial presentation and is most commonly performed due to intractable right- or left-sided heart failure.17,18
Cascade screening of family members, the fifth component of ARVC management, is discussed in the ARVC risk stratification section below.
Arrhythmogenic Right Ventricular Cardiomyopathy Risk Stratification
The approach to ARVC risk stratification has evolved considerably over time. A general guideline to follow is the more severe the disease, as assessed from an electrical and structural perspective, the greater the risk of sustained VA or SCD. This approach is based on a long list of risk markers that have been identified. These risk markers include: previous history of sustained VT or VF; premature ventricular contraction (PVC) frequency; non-sustained VT (NSVT); cardiac syncope; proband status and genetic testing; gender; degree of exercise restriction; and degree of myocardial involvement.
As identified in Orgeron et al., and supported by previous studies by Mazzanti et al. and Piccini et al., a history of VA predicts appropriate ICD therapy for any future VA.19–21 Corrado et al. suggested that the risk for VF is likely to be low in patients with a history of haemodynamically stable VT, but other studies have suggested that haemodynamically significant VT is still associated with an unacceptably high risk for lethal VA and SCD.22 As a result, a history of any VA is a potent risk factor. Current guidelines advise ICD implantation for all patients with ARVC who have had a previous sustained VA.23,24 However, the more critical issue concerns risk stratification in patients who have never had a sustained VA. In the following sections, we discuss some of the most important primary prevention risk markers.
Electrical Instability
Electrical instability is an important risk factor in ARVC risk stratification. PVC burden (more than 1,000 in 24 hours), NSVT or more invasive evaluation with electrophysiology study can all quantify electrical instability. High PVC burden, NSVT and a positive electrophysiology study (defined as VT >30 seconds or haemodynamically significant VA requiring termination) all predict appropriate future ICD firing (for VT/VF). Interestingly, only high PVC burden was found to be predictive of future VF or ventricular flutter (Vfl) events.19,25–29 Only one study, by Corrado et al. in 2010, found that inducibility was not predictive of future appropriate therapies, and cited a positive predictive value of programmed ventricular stimulation (PVS) of 35% for any appropriate ICD therapy, and only 20% for VF/Vfl.30 However, given the abundance of evidence suggesting an association with VA and or appropriate ICD therapy, inducibility should be considered a marker of higher risk ARVC, along with high PVC burden and history of NSVT.
Cardiac Syncope
A history of syncope should raise suspicion for a previously unrecognised VA event. A detailed history of the syncopal event should be carried out to search for clues of cardiac origin (diaphoresis, shortness of breath, palpitations, severe injuries sustained from syncopal event). In 1989, Marcus et al. first recognised that previous syncope was associated with worse outcomes in ARVC, including arrhythmic death.31 Later studies confirmed this and found that syncope at first presentation was not only common but also suggestive of poor outcomes, such as SCD.32,33 One of these studies found that approximately half of patients diagnosed with ARVC after SCD had syncope prior to the event.32 In line with these findings, syncope also predicts VF/Vfl and appropriate ICD therapy.19,30 Syncope (especially when there is a high suspicion of cardiac origin) should be considered as a significant risk factor for poor outcomes in patients with ARVC.
Genetic Testing and Proband Status
ARVC is a disease of desmosomal dysfunction. Eighty per cent of patients have a single copy mutation of the plakophilin-2 (PKP2) gene, but the less common mutation of the desmoplakin gene is associated with significantly higher rates of SCD. Importantly, the rate of first life-threatening arrhythmic event (LAE) in one study was found to be greatest between the ages of 21 and 40 years, measured at 4.0 per 100 person years.24 This suggests that children in families with ARVC should be screened for these mutations in the teenage years, prior to this period of increased LAE risk. Patients with more than one identified mutation are at an even higher risk for not only earlier onset of symptoms, but also for SCD and VA.25 The importance of cascade screening of family members after a proband identification is further supported by increased rates of appropriate ICD therapy in probands versus family members.34 It is likely that the lower rate of ICD therapy in family members is due to earlier initiation of appropriate treatment and exercise restriction.35
Gender
Despite most ARVC mutations being transmitted in an autosomal dominant fashion, male sex is a well-recognised predictor of lifetime arrhythmic risk.36 The pathophysiology of this was originally thought to be attributed to exercise, but the role that sex hormones play in ARVC is becoming well-recognised. Akdis et al. found that higher levels of testosterone in men and lower levels of oestrogen in women were both associated with higher rates of major arrhythmic cardiovascular events in patients with ARVC.37 The underlying pathophysiology is thought to be due to testosterone promoting apoptosis and lipogenesis, while oestrogen inhibits these effects. This possibly explains why regular exercise, which is thought to lower oestrogen levels, is associated with worse outcomes in ARVC.38 Additionally, these findings may provide an explanation for the rare occurrence of ARVC before pubertal years.
Other Risk Factors
Exercise has also been associated with progression and poor outcomes in patients with ARVC. Even ARVC carriers with greater exercise duration were found to be at significantly higher risk for a VA event. This suggests higher penetrance in patients who exercise more.39 This finding was similarly demonstrated in patients with definite ARVC. Athletes with definite ARVC were at higher risk for VA, VA at a younger age, and ICD placement at a younger age.6,40 Wang et al. further investigated exercise restriction and found a dose–response relationship between exercise dose (defined as intensity × duration) and reduction in VA events.14 This benefit is even greater in gene-elusive patients and those with ICDs placed for primary prevention.14 Despite this, the absolute risk of VA in that study was found to be 22% annually.
Another recent study by Gasperetti et al. reinforced these findings by showing a reduction in PVC burden through ‘detraining’ of athletes with ARVC. However, there was no improvement in RV ejection fraction (RVEF) with exercise restriction.40 An additional study by Maupain et al. further supported exercise restriction by finding an increased VA risk in those who exercised more than 6 hours per week.29 All of these findings together suggest that exercise restriction alone is not sufficient to avoid ICD placement, but it should be strongly recommended to patients with ARVC to reduce VA events. Accordingly, the current 2019 Heart Rhythm Society (HRS) Guidelines for ARVC Management recommend that those with ARVC should avoid competitive exercise and high-intensity endurance exercise.10 Exercise intensity is expressed using metabolic equivalents (METs). Low-MET activities such as yoga and walking for pleasure should be considered safe, and even encouraged. However, more intense exercises, such as running, swimming and sports, should be avoided given the deleterious effects of exercise.10 Unwillingness to restrict exercise should be considered during risk stratification and decision-making regarding ICD placement.
Degree of myocardial involvement should be assessed as well. Extensive RV involvement, defined as RVEF ≤45% or ≥2 areas of regional dysfunction, is predictive of appropriate ICD therapy.41 Greater RV dysfunction as measured on echocardiogram is associated with an overall increase in major adverse cardiovascular events (MACEs), with VA unsurprisingly being the most common MACE.42 In addition to RV involvement, LV involvement in ARVC is also recognised.2 An early study by Wichter et al. in 2005 suggested that LV involvement showed a trend towards predicting appropriate ICD therapy.41 More recent studies have provided stronger evidence that LV involvement and dysfunction increase VA risk.29,42–44 Given the progressive nature of ARVC, we suggest that the degree of myocardial involvement is reassessed periodically to re-stratify a patient’s risk.
Decision-making Regarding ICD Placement
ICD placement is currently the cornerstone of treatment after a diagnosis of ARVC. However, the decision to place an ICD should be weighed against the short- and long-term risks of ICD placement. According to the 2015 International Task Force (ITF) consensus statement, Class I indications for ICD placement include a history of sustained VT or VF, severe RV dysfunction (fractional area change ≤17% or RVEF ≤35%), and severe LV dysfunction (LVEF ≤35%; Figure 1).23 However, this ITF consensus statement is less clear regarding ICD placement for primary prevention in ARVC patients. The ITF consensus statement provides Class IIa indications for when ICD placement should be considered in patients with ≥1 major risk factor, such as a history of syncope, NSVT, moderate RV dysfunction (RV fractional area change between 24% and 17% or RVEF 36–40%), moderate LV dysfunction (LVEF 36–45%) or biventricular dysfunction. Class IIb indications for when ICD placement may be considered include T wave inversion in ≥3 precordial leads, male sex, inducibility on electrophysiological study, and proband status.23 These guidelines lack clarity regarding ICD placement for primary prevention in ARVC patients.
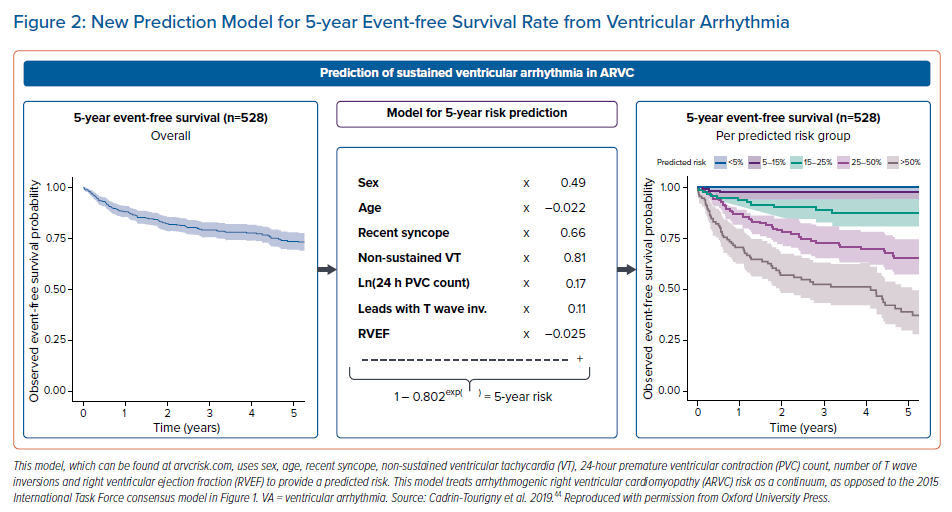
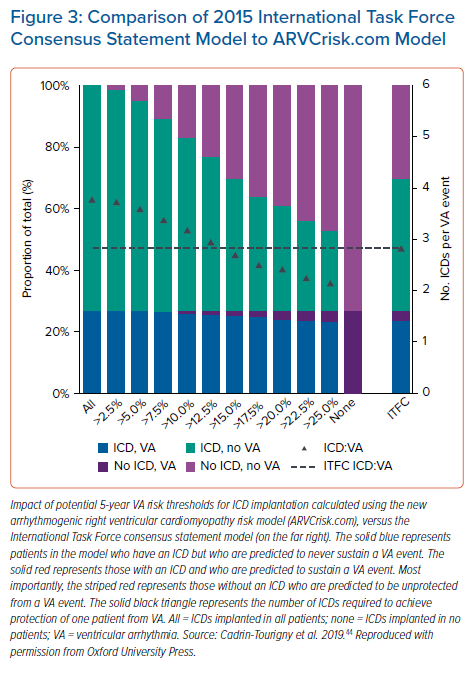
A paper by Cadrin-Tourigny et al. in 2019 attempted to provide more clarity regarding primary prevention ICD placement by creating an ARVC risk calculator (which can be found at http://www.arvcrisk.com).45 This risk calculator uses age at diagnosis, sex, cardiac syncope (<6 months prior to diagnosis), number of T wave inversions, maximum 24-hour PVC count, history of NSVT, and RVEF to provide a predicted risk of sustained VA. Compared with the 2015 ITF algorithm, which treats ARVC risk as high, intermediate or low, the more recent ARVC risk calculator treats VA risk as a continuum. This new algorithm showed similar levels of benefit and protection from ICD placement at a much lower rate of ICD placement (20.6% fewer ICD implantations compared with the ITF algorithm; Figures2 and 3).45
Another study comparing this new ARVC risk score with both the ITF and HRS guidelines found that an ARVC risk score >10% had the greatest net benefit compared with the guidelines.46 This new model provides the physician with a tool with which to quantify an individual patient’s risk and which can supplement clinical judgement during ICD placement decision-making. It is worth mentioning that this new model may underestimate non-classical forms of ARVC, such as biventricular or left dominant forms.47 Notably, this risk calculator also does not include inducibility on PVS, which is one of the discussed risk factors in the present article. Future ARVC risk calculators may include this to provide even more robust models regarding ARVC risk.
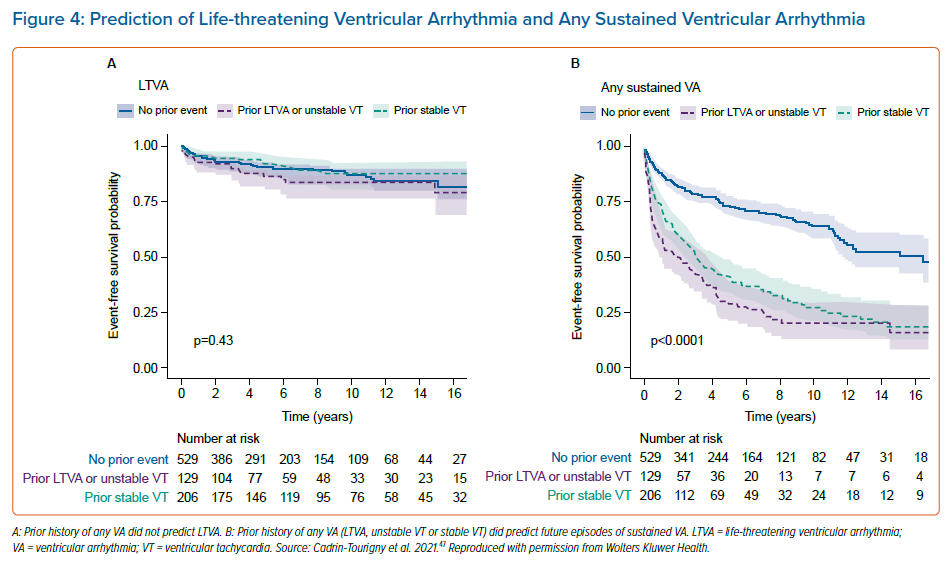
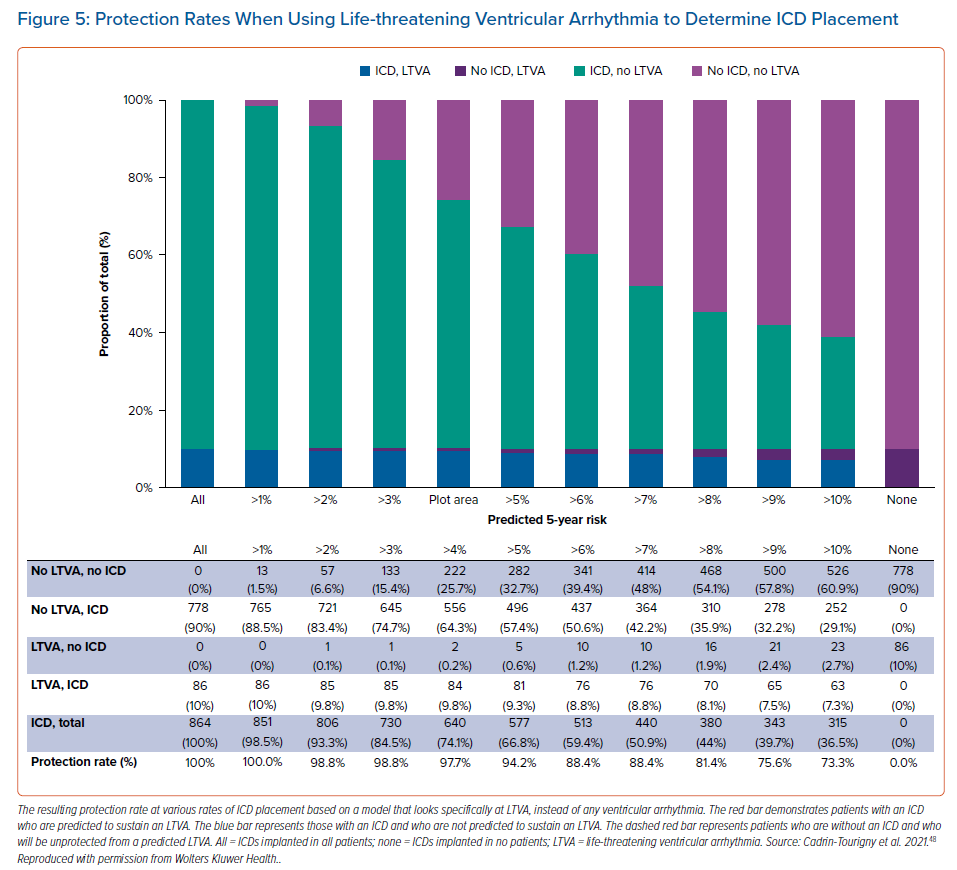
An even more recent study by Cadrin-Tourigny et al. specifically investigated predictive factors of life-threatening VAs (LTVAs) to serve as a closer surrogate marker for SCD risk. That study did not find that prior sustained VA predicted LTVA, but that younger age, male sex, PVC count and number of leads with T wave inversion were predictive of LTVA.48 However, that study did reinforce the predictive value of any previous sustained VA for a future sustained VA event (Figure 4).48 That study also equips clinicians with more data for shared decision-making regarding ICD placement by providing more objective measures of risk and protection rates (Figure 5).48 It may not be unreasonable to forgo ICD placement in those deemed high risk for complications, or in low-risk patients who are hesitant to have an ICD placed. Regardless of these data, current ARVC guidelines recommend ICD placement as secondary prevention in patients with a history of any VA.23.
Conclusion
Risk stratification should be carried out immediately when a diagnosis of ARVC is made. Risk factors predictive of VA and SCD include age of onset, male sex, specific genetic mutation, cardiac syncope, history of VA, degree of myocardial involvement, electrical instability, and exercise restriction. An individualised risk assessment is required to weigh the risks and benefits in the important decision of whether or not to proceed with ICD placement.
Clinical Perspective
- Arrhythmogenic right ventricular cardiomyopathy (ARVC) management consists of a 5-step approach that includes accurate diagnosis, determination of the need for ICD placement, minimisation of ICD therapy, prevention of disease progression and cascade screening of family members.
- ARVC risk stratification is determined by age at presentation, male sex, proband status, history of ventricular arrhythmia, history of cardiac syncope, frequency of premature ventricular contractions and non-sustained ventricular tachycardia, degree of myocardial involvement and exercise plans.
- ARVC risk calculators provide more objective measures of risk stratification and can supplement clinical judgement when weighing the risks and benefits of ICD placement.