










Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a rare inherited arrhythmia syndrome or channelopathy characterised by exercise- or emotional stress-induced bidirectional or polymorphic ventricular tachycardia in the setting of a structurally normal heart and a normal ECG.1 It is estimated to affect 1 in 10,000 people with reported mortality rates as high as 30–50% by the age of 35 years.2–4 This review presents an update on our current understanding of the condition and its clinical implications.
Clinical Presentation
CPVT is a heterogeneous disease that manifests primarily in children, with a mean age of 7–9 years at presentation. It is recognised as an important cause of unheralded sudden cardiac death (SCD) in young people.3,5,6 Although most patients will present before the age of 10; there are some reported cases where the patient has first presented in their 20s and 30s.7 Many people with CPVT will have a family history of SCD at a young age and/or stress-induced syncope or presyncope.8,9
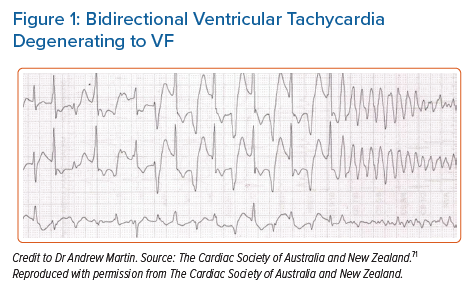
Patients with CPVT can be asymptomatic and only diagnosed as part of family screening. Symptomatic patients usually present with exercise-induced or emotional stress-related syncope accounting for up to 80% of cases.5 A significant subset of patients present for the first time with cardiac arrest due to ventricular tachycardia (VT) or VF (Figure 1).7,10–12 There are few differences based on sex with a tendency towards earlier manifestation in boys compared to girls.7
Patients with CPVT have a structurally normal heart and a normal 12-lead ECG at rest. The characteristic arrhythmias are bidirectional VT or polymorphic VT in the context of a catecholaminergic surge (Figure 1). This may cause haemodynamic compromise that results in syncope. Faster or sustained episodes of VT may degenerate into VF, resulting in sudden arrhythmic death or aborted cardiac arrest. Some episodes may be short-lived and terminate spontaneously, often causing palpitations and/or presyncope.7,13,14
Physical examination of patients with CPVT is often unremarkable. The syncopal episodes at presentation are often considered vasovagal events or caused by neurological triggers. A considerable proportion will be incorrectly diagnosed with epilepsy due to their presentation with seizure-like activity during syncope.5,2 The typical delay between the first syncope and the establishment of the diagnosis has been reported as 2 years.7
Genetic Basis
CPVT principally arises from pathogenic variants in two genes: ryanodine receptor 2 (RYR2) and calsequestrin 2 (CASQ2). RYR2 is inherited in an autosomal dominant manner and accounts for most cases (55–65%).7 Conversely, CASQ2 demonstrates autosomal recessive inheritance and accounts for approximately 5% of cases. RYR2 is a sarcoplasmic reticulum membrane channel protein expressed in cardiac tissues, and CASQ2 is a calcium storage protein in the sarcoplasmic reticulum. Both proteins play a critical role in regulating intracellular calcium. Pathogenic variants in these genes may result in excessive calcium release during diastole that result in intracellular calcium overload and subsequently delayed afterdepolarisation and triggered activity that could induce VT and VF. Other CPVT-associated genes have been reported with variable frequency of disease causation.15 Using the ClinGen gene curation framework, all CPVT reported genes had been reviewed by an expert panel who classified five genes in addition to RYR2 and CAQ2 as having definitive to moderate evidence for disease causation in CPVT (three calmodulin genes – CALM1, CALM2, CALM3, triadin [TRDN] and trans-2,3-enoyl-CoA reductase-like [TECRL]). Three disputed genes (potassium inwardly rectifying channel subfamily j member 2 [KCNJ2], plakophilin 2 [PKP2], sodium voltage-gated channel alpha subunit 5 [SCN5A]) were deemed unrepresentative of CPVT. The remaining reported gene (ankyrin 2 [ANK2]) was too common in the population to be the cause of disease.16
Patients with RYR2 pathogenic variants have similar clinical courses when compared to patients without RYR2.7CASQ2-CPVT may be more severe and more resistant to β-blockers, although this observation lacks comparison with a large series of individuals.5 In patients with no family history of CPVT, sporadic mutations are likely to be the cause. Of note, missense mutations in RYR2 have also been linked to arrhythmogenic right ventricular cardiomyopathy (ARVC-2) characterised by exercise-induced polymorphic VT that does not appear to have a reentrant mechanism, occurring in the absence of significant structural abnormalities.17 This probably represents an early phenotypic misclassification.
Consensus guidelines recommend genetic testing for patients with clinically suspected CPVT identifying the index case in a family (the proband), and provide subsequent cascade screening of family members to detect relatives harbouring the causative variant.18 In a large retrospective study of autopsy-negative sudden death, RYR2 pathogenic variants were the most common molecular diagnosis, and when a rare variant burden analysis (a statistical test of association) was undertaken, RYR2 was the only gene associated significantly with autopsy-negative sudden death.19
Nevertheless, results from genetic testing should be interpreted with caution and require careful adjudication of variant pathogenicity.20 Data from exome sequence collection, such as that by the Exome Aggregation Consortium, showed that approximately 3% of healthy individuals harbour a rare protein-altering variant in the RYR2 gene.21 The lower the clinical pre-test probability for CPVT, the higher the rate of false positive results, emphasising the importance of comprehensive clinical phenotyping before considering genetic testing.4 One study suggested the potential use of phenotype-enhanced variant classification in CPVT using a newly developed diagnostic scorecard to reduce the burden of RYR2 variants of unknown significance (VUS). The study was retrospectively conducted and revalidated in two big centres. The readjudication using amended standards dropped the VUS rate significantly from 48% to 7% in the primary centre with similar results seen in the validation centre. These results illustrate the potential value of incorporating a standardised assessment of phenotypic strength into the variant classification and reporting process.22
Criteria, Diagnostic Studies and Differential Diagnosis
Clinical Criteria
CPVT should be suspected in patients with exertional syncope or syncope occurring during acute emotion. It should also be suspected in patients presenting with aborted SCD triggered by acute emotional stress or exercise. An exercise test may show ventricular arrhythmia in the form of polymorphic or bidirectional VT. According to the most recent international guidelines from the Heart Rhythm Society (HRS), the European Heart Rhythm Association (EHRA) and the Asia Pacific Heart Rhythm Society, CPVT is diagnosed in the presence of a structurally normal heart, normal ECG and unexplained exercise- or catecholamine-induced bidirectional VT or polymorphic ventricular premature beats or VT in people aged under 40 years. The diagnosis can be made in patients (who can be the index case or family member) who have a pathogenic mutation and in family members of a CPVT index case with a normal heart who have exercise-induced premature ventricular contractions or bidirectional/polymorphic VT. Although very rare, the diagnosis can be made in people over 40 years in which case coronary disease should be excluded.23
Diagnostic Studies
The resting ECG is normal. Thus, the primary role of the resting ECG in patients with CPVT is to exclude other possibilities of arrhythmic cardiac syncope in young patients, such as long QT syndrome (LQTS) and Brugada syndrome.24 Sinus bradycardia has been reported in about 20% of CPVT patients.25 Prominent U waves are also observed in a subset and may also be related to altered intracellular calcium handling.26 Supraventricular arrhythmias, which might be catecholamine related, have been reported and may accompany sinus node dysfunction.8
The exercise ECG test is the most useful diagnostic tool in patients with suspected CPVT and it has a primary role in guiding therapy in confirmed cases. During exercise, isolated premature ventricular complexes (PVCs) often present first. As the exercise continues, they usually develop into ventricular bigeminy followed by polymorphic complexes. If the exercise is stopped at this stage, the ventricular complexes are likely to disappear. These exercise-induced ventricular complexes might be the only abnormality observed in some patients with CPVT who are mildly affected by the disease. Characteristically, the heart rate during which the ventricular dysrhythmias occur is between 100 and 130 BPM, which is typically reproducible. The complexity of the ventricular dysrhythmias is likely to worsen as the exercise workload increases. The occurrence of exercise-induced bidirectional VT and 180° rotation of the QRS axis from beat to beat is highly characteristic of CPVT. Some patients, however, will only develop polymorphic VT in the absence of a stable QRS vector alternans.8,27
Although premature ventricular complexes can also be observed in healthy subjects in response to exercise testing, certain features of PVCs during an exercise test can potentially assist in distinguishing CPVT from healthy controls. These features include: a larger number of PVCs, first appearance at higher workload, a left bundle branch block and an inferior axis, bigeminy or trigeminy at peak stress, QRS duration of more than 120 ms, a coupling interval of more than 400 ms, and disappearance in the first minute of the recovery. Of note, a positive exercise test for bidirectional or polymorphic VT is highly predictive of CPVT, however, a negative test is unreliable for ruling out the condition.28,29
Continuous ambulatory monitoring can also reveal arrhythmias typical for CPVT if the sinus rate of the patient exceeds the arrhythmia-inducing threshold during monitoring. Ambulatory monitoring can be useful in young children, who may find that performing a maximal exercise stress test is difficult. Implantation of a loop recorder can also be valuable in selected patients. On the other hand, provocation testing by IV infusion of isoproterenol or epinephrine can help diagnose patients with concealed CPVT who are unable to exercise.30 The test is considered positive for CPVT if three or more beats of polymorphic VT or bidirectional VT are provoked and borderline if polymorphic couplets, PVCs or non-sustained monomorphic VT are induced.31
Family Screening
CPVT has unfavourable clinical manifestations and prognosis. It is therefore essential to expand the evaluation to first- and second-degree relatives to find other potential CPVT cases in a family. Clinically, exercise testing and Holter monitoring are used for screening. Of note, some CPVT patients may have a negative exercise test in early childhood and become positive later in life. Therefore, regular follow-up with repeated exercise stress tests is indicated.
About 50% of the relatives identified by cascade screening as carrying the RYR2 mutation have a CPVT phenotype. Screening of family members by genetic testing is therefore recommended when a pathogenic or likely pathogenic variant has been identified in the proband. Genetic evaluation facilitates diagnosis in silent carriers and allows implementation of preventive pharmacological therapy and a reproductive risk assessment.23,32
Differential Diagnosis
When evaluating a patient with exertional symptoms attributed to ventricular arrhythmias, one should rule out other conditions that might present similarly. Arrhythmogenic right ventricular cardiomyopathy (ARVC) predisposes to VT and SCD in young people; however, it typically presents with an abnormal ECG, monomorphic VT and evidence of structural changes on cardiac imaging.33 In one study, some patients with a clinical diagnosis of CPVT exhibited PKP2 pathogenic mutation which suggests that the progression of the PKP2-dependent electropathy can be independent of structural disturbance and can precipitate exercise-associated presentations prior to the development of an overt cardiomyopathy, clinically mimicking CPVT.34 This phenotypic overlap calls for careful imaging assessment (echocardiogram/cardiac magnetic resonance) in all patients with suspected CPVT. LQTS may present with exercise-related syncope, typically in the LQT1 variant.35 Occasionally, some individuals with Andersen Tawil syndrome, a variant of LQTS, may develop bidirectional VT similar to that of CPVT; however, the presence of QTU prolongation on ECG, syndromic features, the lower risk of sudden death and inconsistency of the relationship of arrhythmia to adrenergic activity distinguish this condition from CPVT.7, 36 Short-coupled torsade de pointes (SC-TdP) may also present similarly. It occurs in the setting of structurally normal heart and normal ECG. However, the onset of the SC-TdP is not related to adrenergic stimuli and is not associated with the typical bidirectional pattern of CPVT tachycardia.32 Another heritable cause of polymorphic VT is familial ST depression syndrome, although its presentation is not related to exercise and some patients will have left ventricular systolic dysfunction.37–39
Risk Stratification and Treatment
The occurrence of cardiac arrest is associated with a higher risk of future arrhythmic episodes. Symptomatic patients with arrhythmic syncope are more likely to develop a cardiac event compared to those who are asymptomatic. Similarly, diagnosis at a young age is linked to adverse outcomes.3 Persistence of complex ectopy during exercise tests may be a marker of adverse outcomes as well.3 Ventricular arrhythmias are usually not inducible at programmed electrical stimulation, making it inadequate for management and risk stratification. Furthermore, the predictive value of inducibility of ventricular arrhythmias by catecholamine infusion or exercise for risk stratification has not been demonstrated.
The role of genetic testing in risk stratification is evolving. The attribution of pathogenicity to a rare variant in the RYR2 gene is challenging, given the high rate of rare, yet benign variants. CPVT-causing variants tend to cluster in functionally relevant domains. Patients with RYR2 variants affecting the C-terminus (CTD amino acid) are at higher risk of life-threatening arrhythmic events independent of clinical presentation and the type of β-blocker used.40 In a retrospective observational study, de novoRYR2 variants were more likely to be located in the C-terminus domain and less likely in the N-terminus domain than in the familial group. In this cohort, the de novo cases presented with an arrhythmic event (syncope or cardiac arrest) at a younger age.41
Lifestyle changes and supportive care are crucial for all patients with CPVT. Furthermore, all patients with CPVT should be treated with pharmacological therapies to reduce the incidence of the primary manifestations of the disease. Non-pharmacological treatments should be considered in selected patients with persistent symptoms and/or exercise-induced ventricular arrhythmias despite optimal medical therapy, and device therapy is recommended for secondary prevention.
Sport and Lifestyle Changes
Due to the nature of the condition, long-term management strategies in CPVT have the objective of reducing adrenergic stimulation. Lifestyle modification, including the avoidance of emotional distress, is advised. A position statement by the European Association of Preventive Cardiology (EAPC) and the European Heart Rhythm Association (EHRA) suggests the following for patients with CPVT:42
- Competitive and intensive leisure time sports are not recommended.
- Low-to-moderate-intensity leisure-time sports may be considered if stress tests show the absence of any ventricular ectopy or arrhythmia when using appropriate treatment, and if the patient is asymptomatic for a minimum of 3 months, including patients with an implantable cardioverter defibrillator (ICD).
- Restrict sports activity to a moderate intensity of 30–60 minutes a day on 3–7 days a week, similar to the general population.
- Carriers of a pathogenic CPVT mutation without an overt phenotype should be managed as patients with manifest CPVT and be restricted to low-intensity sports.
- Follow-up should include stress test and/or continuous ECG monitoring during low-intensity sports to ensure control of exercise-induced ventricular arrhythmias.
- Avoid stressful and emotional situations, dehydration, electrolyte disturbance and hyperthermia.36,42,43
Despite these recommendations, preliminary retrospective analysis suggests that with appropriate treatment, sports practice may be possible if sudden bursts of exercise are avoided.44 Based on previous exercise testing, individualised maximum heart rate limits may be specified. Of paramount importance is that a diagnosis of CPVT can be a burden for patients as well as their families and appropriate psychological support should be considered early on.45
Pharmacological Therapy
β-blockers are the first-line therapy. Current guidelines recommend treating symptomatic patients with β-blockers.20 High-dose nadolol 1–2 mg/kg is preferred to other types of β-blockers as patients on nadolol have lower event rates.46,47 In countries where nadolol is not available, another non-selective β-blocker, such as propranolol, could be used as an alternative. It is important to emphasise that β-blockers should be administered throughout pregnancy in affected women. Asymptomatic patients with positive pathogenic mutations for CPVT and negative exercise tests have lower arrhythmic event rates on long-term follow-up (4.4 per 1,000 person-years), nevertheless, they may be considered for prophylactic β-blocker therapy.32,23
Although β-blockers are the mainstay treatment for CPVT, they are not fully effective in all patients. Van der Werf et al. reported 37% of patients on β-blocker will continue to develop ventricular arrhythmias at 8 years follow-up. Furthermore, the fatality rate on β-blocker has been reported as being between 2.6–9.2%.20,48 Therefore, regular evaluation with exercise tests is mandatory to up-titrate the β-blocker to the maximum tolerated doses, emphasise the importance of strict compliance with therapy and identify those who may benefit from treatment escalation.7
Flecainide effectively reduces ventricular arrhythmias in symptomatic patients with CPVT already on a maximum tolerated dose of β-blocker. The current guidelines recommend the consideration of flecainide in addition to β-blocker in patients with recurrent syncope, ventricular arrhythmias on exercise stress test, or to reduce appropriate ICD therapy.23 The usual daily dose of flecainide ranges between 100 and 300 mg.49,8 A recent randomised crossover trial has shown flecainide to be very effective when taken in addition to β-blocker in suppressing ventricular arrhythmias in patients with CPVT. The authors suggested that titrating flecainide dose depending on plasma trough levels achieved better outcomes.50 In another study, the antiarrhythmic effect appeared to be independent of the specific CPVT genetic subtype.51 Flecainide monotherapy has been reported in some cases as well as a small observational study; however, there are no randomised trials to evaluate its effect on patients who are not also taking β-blockers.52,53 As it stands, flecainide monotherapy is not recommended in patients who can tolerate β-blockers.23
At present, the only measure of the efficacy of flecainide is the burden of ventricular arrhythmia using the exercise test. It is, however, essential to recognise that this was not a reliable predictor of cardiac events in some reports. Therefore, Lieve et al. suggest that the optimal therapeutic response is evaluated by comparing ventricular arrhythmia burden on exercise before and after the initiation of flecainide.54 Flecainide reduced ventricular arrhythmias effectively in patients on maximally tolerated β-blocker therapy with symptoms or ventricular arrhythmias during their exercise test. These results were similar in patients with pathogenic variants in RYR2, CASQ2 and genotypically negative cases. Nevertheless, large studies are needed to fully evaluate the efficacy of flecainide in prevention of cardiac events in the long term.
Data that support the use of calcium channel blockers are relatively scanty. Studies by Swan et al. in 2005, Rosso et al. in 2007 and Katz et al. in 2010 have investigated the efficacy of verapamil in addition to β-blocker in suppressing ventricular arrhythmias in CPVT patients.55–57 The burden of ventricular arrhythmias was significantly reduced in all three studies in the short term; however, verapamil did not prevent events in these groups in the long term.58 Moreover, in all three studies, verapamil did not add any negative chronotropic effect as the mean heart rate was similar before and after verapamil. Therefore, the current guidelines do not recommend verapamil as first- or second-line therapy. Yet, it could be considered in some patients on an individual basis, particularly in carriers of a specific CASQ2 mutation.53,59
Non-pharmacological Therapy
The European Society of Cardiology (ESC) guidelines and the HRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes recommend ICD implantation in CPVT patients who experienced cardiac arrest, recurrent syncope, or polymorphic/bidirectional VT despite optimal medical therapy and/or left cardiac sympathetic denervation (LCSD).23,60 ICD as a stand-alone therapy in an asymptomatic patient with CPVT is contraindicated due to the risk of adrenergic storms.23 Despite guideline recommendations, real-world data suggests a trend towards a more liberal use of ICDs in CPVT patients.12,61 Given the nature of CPVT and its potentially malignant course even with optimal medical therapy, implanting an ICD seems reasonable. However, ICDs are not without their problems.
Early reports suggested that ICDs might be proarrhythmic in CPVT.62,63 Roston et al. concluded in a systematic review and meta-analysis that ICDs effectively terminate VF but are not as effective in terminating VT.61 Recently, Van der Werf et al. studied 136 patients who presented with SCD in whom CPVT was diagnosed, subsequently leading to the initiation of guideline-directed therapy with β-blocker and flecainide with or without LCSD. 58% of the patients had an ICD implanted. After a median follow-up of 4.8 years, a composite outcome of SCD, sudden cardiac arrest, syncope or appropriate ICD shocks occurred in 47% of the patients with ICD and only 15.8% of the patients without ICD. Moreover, inappropriate ICD shocks occurred in 24% and other device-related complications in 29% of the patients. Although limited by being observational data, this study suggests a lack of survival benefit in patients with ICDs; instead, ICDs are associated with a high rate of appropriate and inappropriate shocks along with other device-related complications.64
Another prospective cohort study followed up 216 patients with CPVT on β-blockers only for a mean duration of 7.5 years. Twenty-eight patients (13%) had a life-threatening arrhythmic event; 18 had an ICD already implanted prior to the event, with subsequent zero mortality; and four of the other ten patients (40%) died. Based on these findings, the authors argued that ICD implantation offers important protection in CPVT. However, relevant clinical information was not presented on whether the patients who died had symptoms while on β-blocker or had a normal exercise test on follow-up before they had cardiac arrest. Notably, data on long-term arrhythmic events in CPVT patients on dual therapy (β-blocker and flecainide) with or without LCSD are still lacking.41
Based on the data available and in the absence of randomised trials, it seems reasonable to recommend a conservative approach towards using ICDs in patients with CPVT in line with the current guidelines. Indeed, when an ICD is implanted following a shared decision-making process, the treating physician should seek to minimise the risk of appropriate and inappropriate therapy by optimising medical treatment with or without LCSD. Supraventricular tachycardias are common in CPVT patients and they should be treated promptly as they facilitate ventricular arrhythmias as well as possibly resulting in inappropriate ICD therapies. ICD programming with an extended monitoring period could also help to minimise these risks, aiming to treat only high-rate arrhythmias, such as VF.
Left cardiac sympathetic denervation is a surgical procedure in which the lower two-thirds of the left thoracic stellate ganglion along with T2–T4 ganglia are ablated, resulting in an interruption of the major source of norepinephrine release in the heart.65 LCSD is at least moderately effective in reducing arrhythmic events in CPVT patients according to multiple reports, although long-term data are still needed.6,43,49,66,67 LCSD is not curative and is not recommended as monotherapy for CPVT patients. There are some limitations related to procedural risks depending on the chosen surgical approach whether that is video-assisted thoracoscopic surgery or open thoracotomy. Possible outcomes including failure of the procedure should always be discussed before surgery to manage the patient and the family’s expectations.68 As it stands, LCSD may be considered in patients with CPVT who are symptomatic with recurrent syncope, polymorphic/bidirectional VT, or appropriate ICD shocks despite maximal medical therapy and/or intolerant of or with contraindication to β-blocker.23
Using a Stepwise Approach
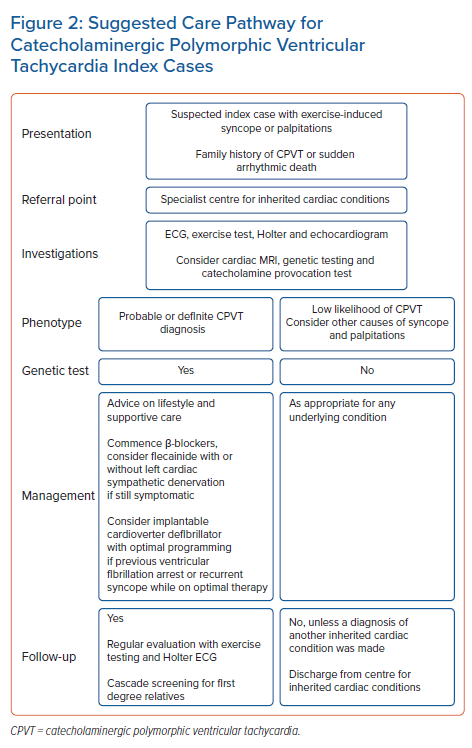
All CPVT patients should be on a β-blocker and be given advice regarding lifestyle changes and avoiding strenuous exercise. Follow-up with exercise testing is mandatory to evaluate the clinical response to therapy. Patients with exercise-induced ventricular arrhythmias in the form of ventricular couplets, non-sustained VT, polymorphic or bidirectional VT should be considered for flecainide add-on therapy. If they continue to have exercise-induced ventricular arrhythmia and/or symptoms, they may be offered an ICD with or without LCSD, although earlier use of the LCSD and avoidance of an ICD is advocated by some. All CPVT cases should be managed by experts in the field in a referral centre.7Figure 2 is a flowchart summarising a suggested care pathway for patients with CPVT.
Knowledge Gap and Future Directions
Despite significant progress in understanding CPVT, there are still uncertainties and gaps in our knowledge. In day-to-day practice, clinicians may make difficult diagnostic and therapeutic decisions without solid evidence. Such difficulties contribute to most clinical dilemmas and must be shared with the patients and their families. Examples include the definition of optimal exercise stress test protocol and the accepted findings on stress tests on follow-up. Furthermore, the background rate of RYR2 rare variants is reported as 3% in a general white population, and functional evidence to support causality is only present in a minority. Interpretation of these rare variants in daily practice remains a significant source of uncertainty and discomfort, particularly when discussing test results with bereaved families.6 Indeed, many patients with CPVT remain variant-negative; therefore, future discovery of genes and gene modifiers will continue with a particular focus on genome-wide association studies.
Lifestyle changes and the ‘acceptable’ level of exercise in probands and asymptomatic affected relatives is another clinical dilemma frequently faced by clinicians. On the management aspect, although nadolol is not available in many countries, there is a lack of data on the use of other non-selective β-blockers in symptomatic patients. On the other hand, in asymptomatic patients with RYR2 pathogenic variants identified on family screening, the safety of not treating with β-blockers has yet to be studied fully. Moreover, studies are required to evaluate the role of more proactive treatment in some high-risk groups (for example, patients with RYR2 variant affecting the C-terminus and those with homozygous variants in the CASQ2 gene). More work is needed to examine the role of electrophysiological testing and catheter ablation in atrial and ventricular arrhythmias in refractory CPVT cases. Finally, gene therapy is a promising new therapeutic option for CPVT.69,70
There is a need for more comprehensive risk stratification tools to aid the management of CPVT patients coupled with interventional randomised trials to evaluate existing and novel therapeutic options. However, CPVT has a low prevalence, and more referral centres need to collaborate to address these priorities. Great examples of the outcomes that can be achieved in this type of collaboration have been demonstrated by the multicentre observational registry that was established in 2014 with future findings expected.47,64
Clinical Perspective
- Catecholaminergic polymorphic ventricular tachycardia (CPVT) is an inherited arrhythmia syndrome that is characterised by stress-induced bidirectional and/or polymorphic VT.
- Exercise tests play an important role in the diagnosis of CPVT as well as monitoring the response to medical therapy in affected patients.
- β-blockers remain the cornerstone therapy for CPVT. The addition of flecainide often further reduces the incidence of arrhythmic events in symptomatic patients.
- Lifestyle changes including avoidance of emotional stress as well as avoidance of competitive and intensive leisure-time sport are recommended.